

Translate this page into:
Screening, molecular simulation & in silico kinetics of virtually designed covid-19 main protease inhibitors
⁎Corresponding author. MMyAlzahrani@imamu.edu.sa (Mohammed AL-Zharani)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Coronavirus (covid-19) infection is considered to be deadliest ever pandemic experienced by the human being. It has very badly affected the socio-economic health of human and stuck the scientific community to think and rethink about its complete eradication. But due to no effective treatment or unavailability of vaccine the health professional could not show any significant improvement to control the pandemic. The situation needs newer molecule, vaccine or effective treatment to control covid-19 infection. Different target in viruses has been explored and proteases enzymes were found to be therapeutically effective target for the design of potential anti-covid-19 molecule as it plays the vital role in viral replication and assembly. Structure-based drug design was employed to discover the small molecule of anti-covid-19. Here we considered the small library of naturally occurring polyphenolic compounds and molecular docking, Molecular dynamics (MD) simulations, free binding energy calculation and in-silico ADME calculations to identify the newer HITs. Based upon their score the two molecules were identified as promising candidate. The docking scores were found to be −7.643 and −7.065 for the HIT1 and HIT-2 respectively. In MD simulations study the RMSD values were found to be 4.3 Å & 4.9 Å respectively. To validate these results MM-GBSA was performed and their binding free energies were computationally determined. The prime energy values of identified HITs (−13412.45 & −13441.8 kJ/mole) were found to be very close proximity to reference molecule (−13493.05 kJ/mole). Then in-silico ADME calculations were performed to calculate the drug likeliness identified HITs. BY considering all the values comparative to reference molecule and obtained in-silico pharmacokinetic properties of identified HITs we can suggest that HIT-1 and HIT-2 would be the most promising molecules that can inhibit the main protease enzyme of covid-19. These two molecules would become the potential drug candidate for the treatment of covid-19 infections.
Keywords
Covid-19
Protease inhibitor
Phenolic compounds
HIT
MD simulation

1 Introduction
The newly reported covid-19 illness in China's Wuhan area has infected billions of human beings and killed hundreds of thousands of human beings at same point of the world (Singhal, 2020). In March 2020, World Health Organization (WHO) declared that covid-19 is a pandemic. The range of instances suggested and deaths related to covid-19 has been gradually rising, wreaking havoc at the socio-monetary fitness of each advanced and developing countries (Mahase, 2020). The many covid-19 viral strains and mutations have prompted the globe and global fitness device to rethink a way to cope with or comprise the existing pandemic crisis. The signs are different in every case reported by diverse sorts or mutated viruses, which makes it harder for scientists and physicians to diagnose and deal with the patient. Common cold, fever, extreme cough, hassle or shortness of breathing, and lack of flavour or odour were recorded in covid-19 patients (Yuki et al., 2020), but those signs can be lacking or found in diverse mutant viruses (Javorac et al., 2020; Jin et al., 2020). The epidemiological survey stated the higher spreadability of covid-19 around (2–2.5 %) and lower (5 %) fatality compared to earlier reported Severe Acute Respiratory Syndrome (SARS) 1.7–1.9 % & 9.5 % and Middle East Respiratory Syndrome (MERS) < 1 % & 34.4 % spreadability and fatality rate respectively (Ahmadzadeh et al., 2020, Organization, 2019; Hilgenfeld, 2014). Nowadays plants derived natural products are gaining importance since they provide small & novel druggable compounds and inspired the researcher to focus on such compounds & make a considerable contribution to human health and well-being. People prefer natural antioxidants for safety, efficacy, cultural acceptability, and lesser side effects. Hence, we thought to identify the compounds from natural polyphenols class. During the primary week after infection, the covid-19 patient's RT-qPCR illustrates the best virus load (Chakraborty et al., 2020a,b; To et al., 2020). Old age and patients with co-morbidities are extra susceptible or susceptible to excessive contamination motion and a large threat of mortality, in step withcovid-19 affected person cohort research (Zhao et al., 2020, Zhou et al., 2020). Multiple organ failure, respiration collapse, acute respiration misery syndrome, sepsis, coronary heart failure, and septic surprise had been all located on this affected person's cohort study. Following the outbreak, some of current medicine applicants have been examined as drug repurposing applicants for potential use in covid-19 infection (Chakraborty et al., 2020a,b). More than 570 therapeutic applicants are being tracked for drug improvement under the covid-19 treatment accelerated program (CTAP) and controlled through the Food and Drug Administration (FDA) (Keretsu et al., 2020; Zhou et al., 2020). According to latest findings, drugs that concentrate on viral replication mechanisms are receiving a number of hobby for potential remedy in covid-19 (Biering et al., 2021). Various antiviral drugs targeting different stages of viral life cycle have been advanced into numerous degrees of medical trials. Covid-19 main protease enzymes are crucial part in the lifecycle of viruses, which are responsible of cleaving polyproteins and keeping viral replication stable (Gioia et al., 2020). Another protease is encoded through infectious viruses, and it performs a crucial element within the viral lifecycle (Roe et al., 2021). As a result, those proteases (3Clpro or Mpro) offer a therapeutically assured target for a powerful viral contamination therapy (Roe et al., 2021). The purposeful polypeptides pp1a and pp1ab, that are required for transcription and replication, have been launched through 3CLpro's conserved catalytic activity. The covid-19 enzyme is liable for encoding those polypeptides. The crucial cleavage interest of the 3CLproin viral replication pathway, in addition tothe shortage of a human counterpart, made it an exciting target for anti-covid-19 medicinal drug improvement. Various researchers have checked out this target, but the query of its important fee as an antiviral drug improvement has but to be answered, and in addition have a look at is needed (Hilgenfeld, 2014; Liu et al., 2020a; Amin et al., 2021; Amin et al., 2021). Moreover, HTVS and computer aided drug design have been proven to powerful tool to virtually design potential inhibitor against the targeted enzyme or receptors (Usman et al., 2018; Mohd Siddique et al., 2020; Siddique et al., 2018). The present study aimed a structure-based design and discovery a potential novel inhibitor which can be used for treatment of corona infection. Covid-19 main protease enzyme was selected as a target and small library of naturally occurring phenolic compounds was used to perform the virtual screening. Co-crystallized structure of covid-19 main protease enzyme was downloaded from the protein databank (PDB) and its potential to select for the computational work was checked and it displayed in Fig. 1. The obtained values as displayed in Fig. 1 suggested that this co-crystallized structure is suitable for using in-silico calculations. The obtained values and results suggested that the covid-19 protease cocrystal structure with PDB id 6LU7 was technically fit to use for computational work.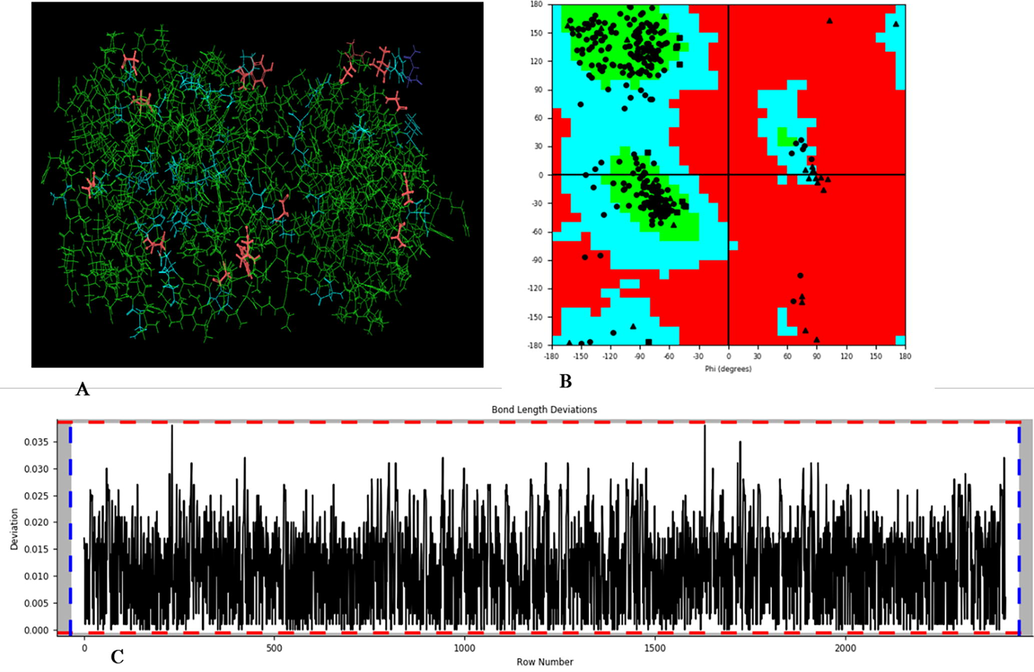
A & B: 3D view of 6LU7 protein analysed for Ramachandran plot, green colour favoured region, cyan colour allowed region and red colour disallowed region. C; RMSD deviation present between each amino acid residues.
2 Material and methods
The preliminary virtual screening of small library of phenolic compounds was carried out using the Schrodinger LLC suite's Maestro (Schrodinger, 2011). Desmond V3 was used for performing the molecular dynamics simulation (Bergdorf et al., 2015). From the openly available (PDB) the co-crystallized structure of COVID-19 main protease (PDB id 6LU7) (Liu et al., 2020b), was retrieved (https://www.rcsb.org). The H bonds were absent from the downloaded raw protein, and certain residues were missing, which were restored using the protein preparation wizard to prevent any unwanted physicochemical constraints. All of the operations were accomplished using the software stated above, which was installed on an Ubuntu operating system 1 TB machine. (Intel (R) C™ i7-8700 CPU 3.20 GHz, 3.19 GHz, 16 GB RAM 1 TB).The typical work flow diagram of proposed is depicted in Fig. 2.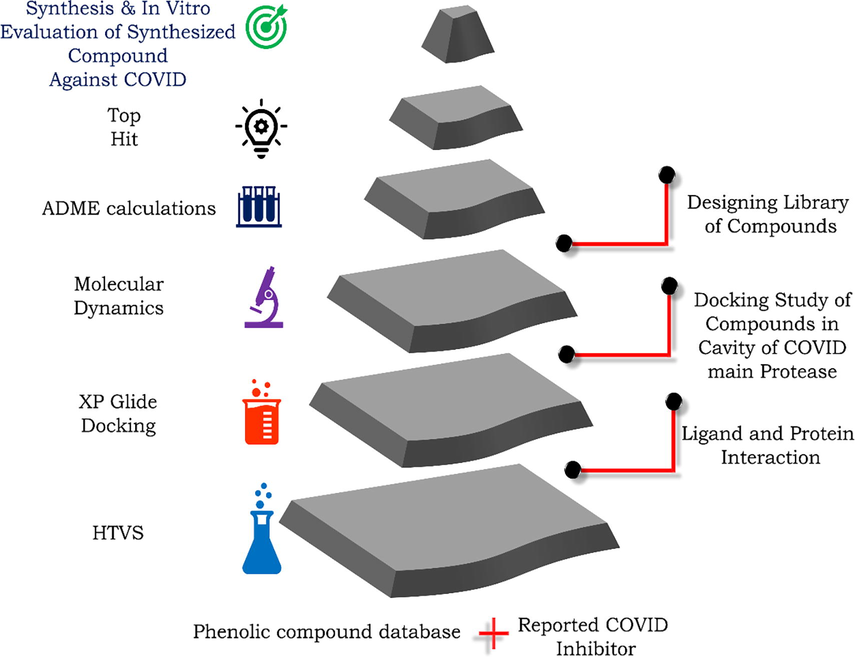
Methodology adopted for the design and discovery of small molecule inhibitors against the COVID main protease.
2.1 Molecular docking studies
The molecular docking studies were performed by using the glide module of Schrodinger tool. The molecules and protein were prepared prior to its use.
2.2 Receptor grid generation
The receptor grid is a cluster of active site residues where a ligand should bind and control the activity of a protein's enzyme. Grid generation was done without any force using Glide's Receptor Grid Generation wizard with default partial cut-off (0.25) and scaling factor (1.0). The site was determined using published literature, and the grid was generated using the centroid of the selected residues.
2.3 Docking
The ligprep of the glide module is used to make numerous conformers and isomers of a screened ligand. There were a total of 32 tautomers and stereoisomers generated. For energy minimization, the OPLS 2005 force field was used, and ligands were subsequently desalted. The glide program was utilized to dock the ligand using the generated gird file with active site residues. The default scaling factor (vdW) was set at 0.8 and the potential charge cut-off was set to 0.15. Importing an xpz file into the XP pose viewer tool was used to analyse the findings.
2.4 Molecular dynamics simulations
The ligand–protein complex at the target site was verified under physiological conditions using an MD simulation study. It was accomplished with the help of a Desmond V3 module operating workstation. The system builder function of the Desmond module integrated the docked protein–ligand complex and utilized it to generate an orthorhombic simulation box. It was developed using a Simple Point-Charge (SPC) explicit water model with a 10 Å distance between the solvent and protein surfaces. This solvated system was neutralized, and physiological salt content of 0.15 M was maintained. MD simulations were conducted using this equilibrated system. The MD simulation was run at 310.15 K temperatures for 100 ns at constant temperature and constant pressure (1.0 bar). Following the successful MD run, the simulation interaction analysis tool was utilized to examine the results obtained by the CMS. During the simulation process, 1000 frames were used to build the MD trajectory, however, only the initial protein backbone frames were aligned to investigate the ligand–protein complex's stability. The RMSD, RMSF, and interaction plots were employed to understand the stability of complexes.
2.5 Molecular mechanics generalized born surface area (MM-GBSA) screening
The results obtained by the HTVS study were re-scored by using the MM-GBSA screening. This is used to check the credibility of preliminary virtually screened results. This was simply done with the 10 best pose of top scoring molecules. The ligand–protein complexes were used to calculate the binding free energy. The complexes were subjected to the prime module of Schrodinger tool with default parameter for such calculations. The molecules were arranged in order of their least binding energy scores (Mali and Chaudhari, 2018).
2.6 In-silico ADME prediction
ADME drug likeliness qualities are such a decisive part of the new drug development process as the numbers of molecules are pulled from the market due to inadequate pharmacokinetic profiles. We used QikProp v3 (Schrödinger, 2005) to predict ADME properties. We used various parameters such as molecular weight, human oral absorption, H bond donor and acceptor, Lipinski's rule of five, and predicted aqueous solubility to calculate the in-vitro and in-vivo drug likeliness properties, as these are important determinants of how the body will react with any external molecule. We considered the identified HIT to be adopted into subsequent phases of drug development processes if all of them come under the same or normal values.
3 Results
Covid-19 main protease 3Clpro enzyme plays a crucial role in viral life cycle process by cleaving the polypeptide required for replication process. Owing to its important role for viral replication, it can be targeted for design and discovery of anti-covid-19 molecule. Here also we consider the covid-19 main protease enzyme for the discovery of small molecule inhibitors of SARS covid-19. Recently, co-crystal structure of 3ClproSARS Cov-19 enzyme with inhibitor N3 was reported by Jin et al. This co-crystallized structure helped us to perform the computational work, analysis of results and proper execution of in-silico data. The CADD drug design is considered as first stage of drug discovery process where the in-silico experiments is performed and based upon the scoring function of molecules, the best one is identified as a suitable molecule that can be taken into further stages of drug design and discovery process. But many promising drug candidates have been failed in later stages of drug discovery process due to less credibility of in-silico results and poor pharmacokinetic profile of drug. This failure leads to the huge loss of financial and time for scientist and an organization. Hence, more validated in-silico data is needed to execute any of the molecule for further study. The dual approach based generated in-silico data can overcome these problems arrived at the later stage and failure of drug discovery process. Here, we also performed the virtual screening small library of natural phenolic compounds against the 3Clpro enzyme. This virtually screened result was validated by the MM-GBSA based binding free energy calculation. Then the stability of drug ligand complex was determined by molecular dynamics simulation and different types of interactions retained after the 100 ns run of MD simulations study was analysed. The Drug likenesses of identified HITs were also calculated to check the in-vivo pharmacokinetic profiling of identified HITs.
3.1 Molecular modelling studies
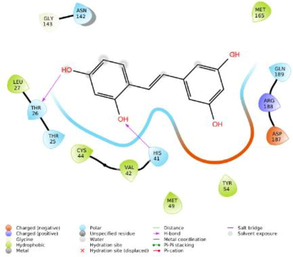
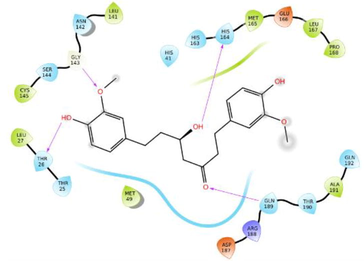
The glide software used for docking study was validated by extracting and redocking of internal ligand binds with the crystal structure of protein. The RMSC value of cocrytalized molecule was found to be 1.869 Å. The previously reported potent covid-19 main protease enzyme inhibitor Ebselen was used as a reference molecule, and the results obtained were compared and analyzed with reference to Ebselen molecule (Wang et al., 2020).The ligand–protein predominant binding modes were predicted by molecular docking studies. Furthermore, structure based docking screening of small dataset of natural phenolic compounds with and COVID main protease enzyme (PDB id 6LU7) was performed. Based upon the docking score molecules were arranged considering the fact that docking scores give an idea about the binding affinity between the ligand and target protein once it docked successfully. The top scoring molecules and different types of interactions obtained in the preliminary screening was summarized in Table 1. These interactions were divided into polar H bond, hydrophobic interactions, polar interactions and π-π interactions. The Fig. 3 displayed the 3D interactive diagram of reference molecule and identified HITs. The analysis of docking results revealed the top scoring molecules and reference molecule, it was found that all the molecules occupied the same active site pocket as that of reference molecule (Ebselen). Ebselen displayed the polar H bond with Gly143 and hydrophobic interactions with Met165; Met49; Cys145; Leu27 active site residues. The H bond might be responsible for the active binding and locking of the molecule while hydrophobic and polar interactions stabilize the ligand–protein complex. From the identified top HITs here, top two compounds were considered for discussion and the data of remaining six compounds were tabulated in Table 1. The phenolic compound 1 and 2 (in Table 1) occupied the same binding pocket with similar fashion with reference molecule and displayed the best docking score −7.643 & −7.065 respectively which was more than the reference molecule. Even the molecular interactions between protein and ligands were also found to be much better than Ebselen. The HIT-1 displayed the seven polar H bond with Thr25, Thr26, Ser14, Asn142, His164, His4 and Gln189 compared to Ebselen which formed only one. Thus, this molecule could bind the more strongly with protein compared to reference molecule because of the seven different H bonding. Similarly HIT-2 displayed the two polar H bonds with His41 and Thr26 residue. Both the molecule did not leave the active binding pocket and found to overlay at the active site only. The HIT-1displayed the hydrophobic interactions with Met49, Ala191, Leu141, Met165, Pro168, Cys145 and Leu27 active site residues and it can be predicted that these hydrophobic interactions and polar interactions with HIT1 could stabilize the docked ligand–protein complex. The HIT-2 displayed the hydrophobic interactions with Leu27, Cys44, Val42 & Tyr54 while polar interactions with Thr26, Thr25, His41, Asn142 and Gln189 respectively. Even though HIT-1 and HIT-2 formed the more interactions with active site residues compared to Ebselen but they were found to devoid of π-π stacking interaction which was found with Ebselen molecule. The other top identified HITs were also displayed the crucial interaction with active site residues as listed in Table 1.
Molecule
Molecule structure
Docking score
Type of interactions
Active site residues
Reference molecule

Polar H-Bond
Gly143
Hydrophobic
Met165; Met49; Cys145; Leu27
Polar interactions
Thr25, Thr26; Ser144; Asn142; His164; His41; Gln189
π-πstacking
His41
HIT-1

−7.643
Polar H-Bond
Thr26; Ser46; Leu141; Gln166; Gln189; Thr190
Hydrophobic
Met49; Ala191; Leu141; Met165; Pro168; Cys145; Leu27
Polar interactions
HIT-2
−7.065
Polar H-Bond
His41; Thr26
Hydrophobic
Leu27; Cys44; Val42; Tyr54
Polar interactions
Thr26; Thr25; His41; Asn142; Gln189
HIT-3
−6.738
Polar H-Bond
Thr26; Gly143; His164; Gln189
Hydrophobic
Leu141; Cyc145; Leu27; Met49; Ala191; Pro168; Leu167; Met165
Polar interactions
Asn142; Ser144; Thr26; Thr25; Gln189; Thr190; Gln192; His164; His163; His41
HIT-4
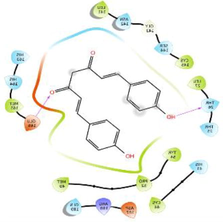
−6.685
Polar H-Bond
Thr26; Glu166
Hydrophobic
Leu141; Cyc145; Leu27; Tyr54; Pro52; Met49; Met165; Cys44
Polar interactions
His163; His164; Gln189; His41; Thr25; Thr26; Ser144, Asn142
HIT-5
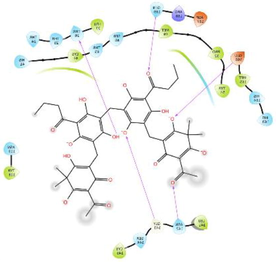
−6.662
Polar H-Bond
Thr26; Gln189; Glu166; Asn142; Gly143
Hydrophobic
Met49; Pro52; Tyr54; Met165; Leu27; Cys44; Leu141; Cys145
Polar interactions
Glu189; Hs164; His163; Asn142; Ser144; Asn119; His41; Thr45; Ser46; Thr26; Thr25; Thr24
HIT-6
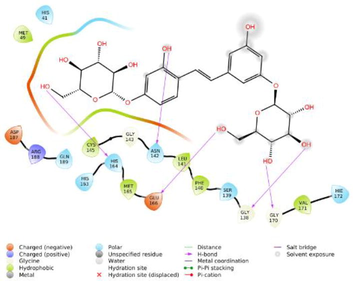
−6.638
Polar H-Bond
Asn142; His164; Glu166; Gly138; Gly170
Hydrophobic
Met49; Cys145; Met165; Leu141; Phe140; Val171
Polar interactions
Hie172; Ser139; Asn142; His164; His163; Gln189; His41
HIT-7

−6.619
Polar H-Bond
Thr26; Gly143; Ap187
Hydrophobic
Leu27; Tyr54; Val42; Pro52; Cys44; Met49; Met165
Polar interactions
Asn142; Thr25; Thr26; His41; Gln189
HIT-8
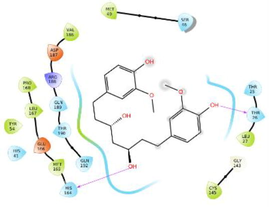
−6.603
Polar H-Bond
Thr26; His164;
Hydrophobic
Leu27; Cys145; Met165; Leu167; Tyr54; Pro168; Val186; Met49
Polar interactions
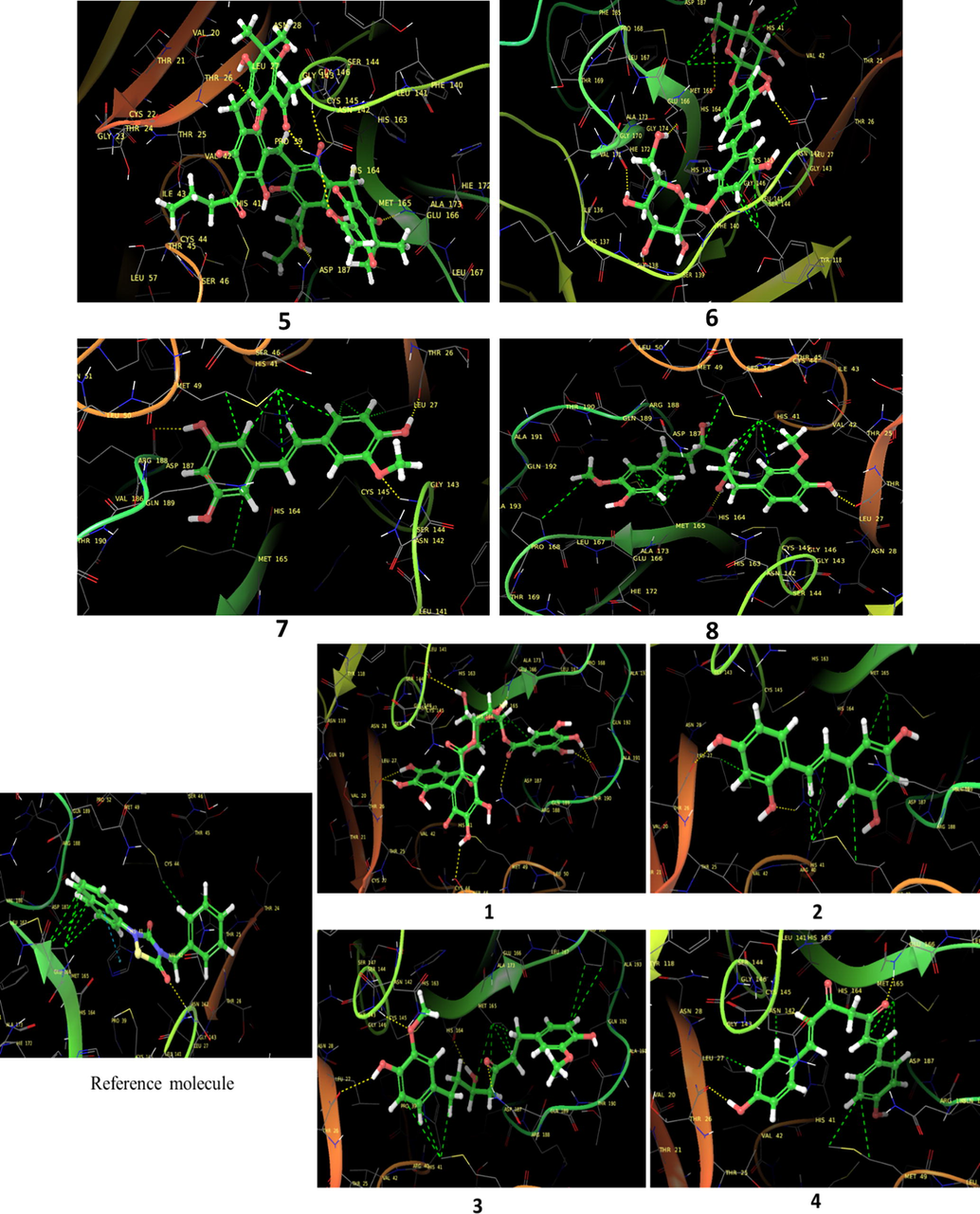
3D overlay of identified top HITs from 1 to 8 at the active site of COVID main protease enzyme. Yellow dash line indicate the polar H bonding interactions between the ligand atom and active site residues whereas green line displayed the hydrophobic interactions.
3.2 Molecular dynamics simulation studies
The MD simulation studies were performed to determine the biophysical interactions between the ligand and protein atoms. In this study the protein and ligand complex were allowed to run for the specific period of time. The study was also aimed to validate the molecular docking results and to check the stability of complex for the 50 ns run of trajectory. Here also we used the active drug-ligand complex for HIT-1 and HIT-2was subjected to MD simulations studies. Based upon the RMSD and RMSF values obtained the stability of ligand–protein complexes for both the molecules were analysed. The various interactions found in ligand–protein docked complexes were also checked whether they are retained or not over entire run of 50 ns trajectory. The time percentage of interactions were also measured and analysed. The binding free energies of both the drug-ligand complexes were also calculated and depending upon their energies the molecules were ranked. The RMSD and RMSF values of ligand and protein backbone were calculated and they are depicted in Fig. 4. The RMSD calculations were performed for the determination of stability of the complex. The RMSD values for HIT-1 and HIT-2 protein complexes were found to be 4.3 Å & 4.9 Å respectively indicating stability of both the complexes. The RMSD value of HIT-1 suggested that the complex was stable for the starting 15 ns after this the complex was slightly deviate with 2.1 Å and it maintained the stability at 3 Å till 40 ns. During the last 10 ns the ligand deviated and maintained the complex stability at the 7 Å. Similarly, HIT-2-protein complex was deviated after the 7 ns after its initial stability and it maintained the stability at 5.4 ns. Hence, we can say that these two complexes were stable throughout the entire MD simulation run of 50 ns. The flexible and rigid site in the protein was determined by root mean square fluctuations, if the molecule fluctuations from the protein site, the molecule may lose binding and stability of complex will be reduced. Thus, we can consider that such molecule with higher fluctuation will not be potential candidate against the selected target. The identified HIT-1 and HIT-2 displayed the very low RMSF values as shown in Fig. 5. By considering the RMSD and RMSF values, it was hypothesized that the identified HITs will not deviate and fluctuate from the active site of covid-19 main protease enzyme. If so, then the ligand–protein was quite stable and the interactions observed in molecular docking studies was also investigated after the entire run of MD trajectory to predict the stability of the complex. If the complex can be retained the observed interaction after the MD simulation run, it will be considered that the complex would be more stable and could produce the inhibitory conformational changes incovid-19 main protease. The RMSF value and ligand interactions with different active site residues for both identified HITs were depicted in Fig. 4 (1b and 2b). The % interactions observed between ligand and active site residues were displayed in Fig. 5 (1a and 2a). The HIT could able to maintain its two polar H bonding with Thr26 for 41 % time period over the entire MD run. The molecule also maintained the different hydrophobic interactions and polar interactions which were observed in molecular docking studies as shown in Fig. 5 (1b and 2b). Two polar H bonding were observed in molecular docking studies of HIT-2 with the active site residues His41& Thr26. But during the MD simulations the molecule formed the polar H bonding with other four different amino acids including Glu189, Glu166, CYs44 and Tyr54 for the period of 58, 57, 68 & 80 % time of entire MD simulation run. Hence, by observing these interactions and comparative analysis with the docked complex and after MD run interactions, we can suggest that these two identified HITs would form the stable complex with covid-19 main protease enzyme.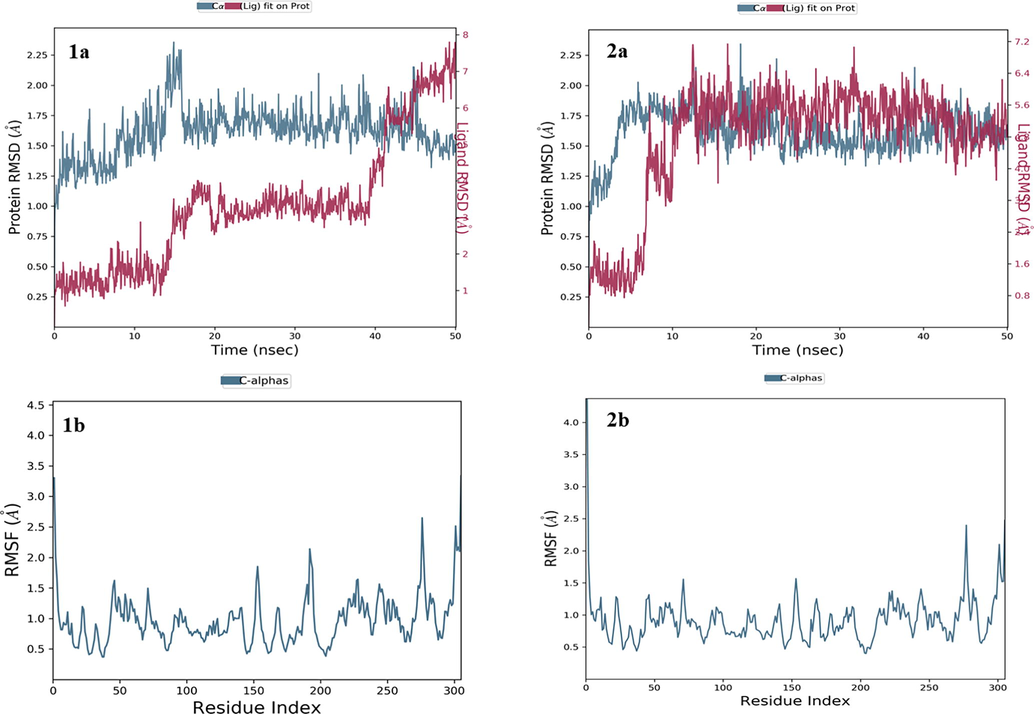
Structural changes (RMSD) over the entire trajectory run of 50 ns upon binding of ligands (1a: HIT1; 2a: HIT2) and Fluctuations of residues (RMSF) at the active site of on binding of ligands (1a: HIT1; 2a: HIT2) Corona virus (PDB id: 6LU7).
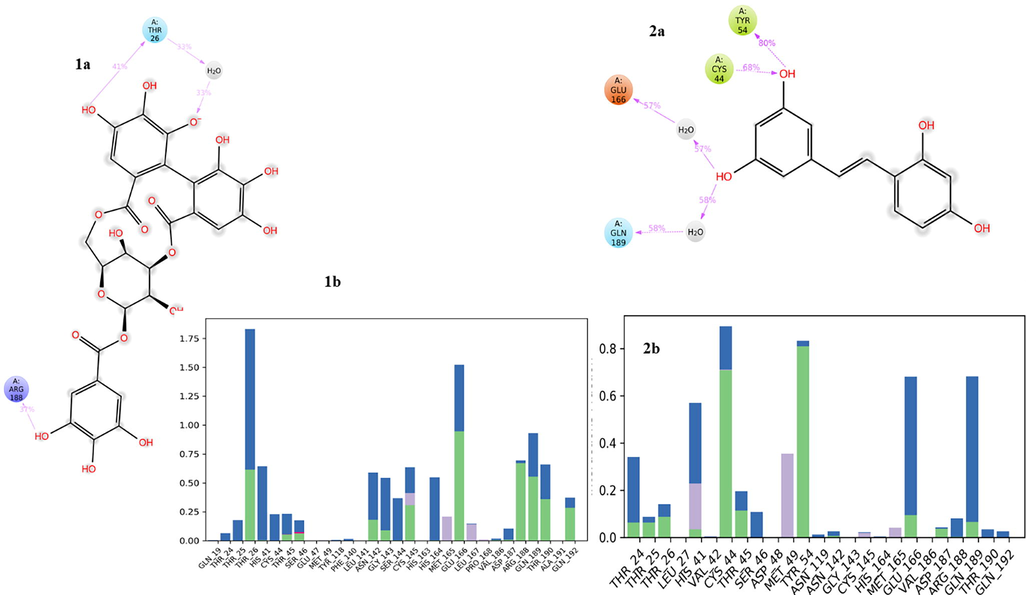
Protein ligands contacts (1a: HIT1; 2a: HIT2) and protein ligand contacts histogram (1b: HIT1; 2b: HIT2) of identified HITs at the active site of COVID Mpro enzyme over the entire run of 50 ns.
3.3 Binding free energies (−ΔG)
The top HIT-protein docked complexes were subjected to MM/GBSA calculations and results obtained were summarized in Table 2. The Prime Energy (−ΔG) (kJ/mole) thus obtained for reference molecule and identified HITs were compared and analysed. The energy values of identified HITs (−13412.45&-13441.8) were found to be very close proximity to reference molecule (−13493.05). Then the H bonding potential of the molecules were also analysed and found to be identical with reference molecule as shown in (Table 2). The charges on ligand–protein complexes were also found to be similar and other properties were also comparable with reference molecule. We know the reference molecule is a potent inhibitor of covid-19 main protease. Its, in vitro inhibitory potential has been published. Now if we get the values of identified HITs identical with this molecule then we can compare HIT-protein docked complex with Ebselen-protein docked complex. The identical values suggested the identified HITs could cause the conformational changes incovid-19 main protease as that of reference molecule and produce the inhibitory effect. Thus, it would be the most promising candidate against the COVID-19 main protease enzyme.
Sr No.
Molecule
Prime Energy
(−ΔG) (kJ/mole)Prime H Bond
Prime Coulomb
1
Reference
−13493.05
−101.59
−10026.37
2
HIT-1 Corilagin
−13412.45
−100.09
−9936.31
3
HIT-2Oxyresveratrol
−13441.8
−100.47
−9958.57
4
HIT-3Hexahydrocurcumin
−13435.92
−102.1
−9694.31
3.4 In-silico ADME calculations
Poor pharmacokinetic profiling of drug candidate may lead to failure of drug discovery process. Even many promising drug candidates failed in later stages of drug discovery processes. This causes the huge time and financial loss. Therefore, early-stage prediction and determination of pharmacokinetic properties is considered to be useful for drug discovery processes, as one can predict the drug likeliness of molecule. Here also the identified HITs with reference molecules were subjected to in-silico prediction of ADME properties by using Qikprop utility of Schrodinger tool. The obtained results were tabulated in Table 3. The different parameters including molecular weight, lipophilicity, hydrophilicity, H bond donor, H bond acceptor, Lipinski’s rule of five, partition coefficient, log P, % oral absorption and blood brain barrier crossing related physicochemical properties were studied. The values obtained indicated the identified HITs were absorbed well and can be distributed to whole body by considering its logP and lipophilicity. The HIT-1 was found to violated the 3 Lipinski’s rule of 5 (Ro5) and it’s percent oral absorption was also found to be very low. The other two HITs did not violate the Lipinski’s Rule of 5 suggesting about the druggable properties of these molecules. The molecules did not cross the blood brain barrier and hence could not produce the CNS related toxicities. By using these values, we determined the pharmacokinetic and druggable properties of identified HITs. Maximum drugs are metabolized by different forms of CytochromeP450 enzyme present in our body. The metabolic behaviour of specific CYPs isoforms with respect to different metabolic sites present in the identified molecules could give crucial information about the pharmacokinetic and pharmacodynamics profile of identified HITs. Amongst different isoforms of CYPs, CYP3A4 was found to be expressed in 30–40 % of total CYPs counts and responsible for the metabolism of many drugs. We used the SMARTCyp (Zaretzki et al., 2013) online free server to find out the potential metabolic present in the identified HITs and results obtained are depicted in Fig. 6.From HIT-1 C-28 was considered to the best site for metabolism by CYP3A4. Followed by this C-27 ad C-32 was most prone to metabolism. Similarly, for HIT-2 C-10 was found to be most suitable position where the metabolism by CYP3A4 is possible. After this C-9 the C-6 were found to be second and third preferential position for metabolism. While forCYP2D6 & CYP2C9 the first metabolic site was found to be C-14 and the second and third positions remain same.
Sr No.
Parameters
Reference
HIT-1 Corilagin
HIT-2Oxyresveratrol
HIT- 3 Hexahydrocurcumin
1
HB Donor
0
11
4
2
2
HB Acceptor
3.5
17.85
3
5.7
3
LogPO/W
4.285
−3.052
1.296
3.605
4
logBB
−0.150
−5.83
−1.773
−2.054
5
#Metabolism
11
4
9
6
% Human oral Absorption
100
40.346
70.575
90.978
7
PSA
56.725
325.167
88.358
105.478
8
Ro5 Violation
0
3
0
0
9
Ro3 Violation
0
0
0
0
10
Molecular Weight
334.39
634.46
244.246
374.433
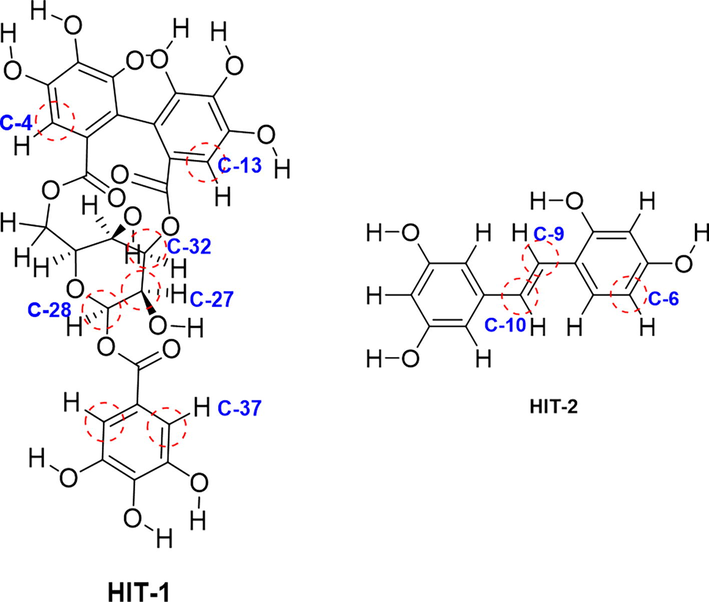
Depiction of different metabolic site in identified HIT-1 and HIT-2, predicted by SMARTCyp.
4 Discussion
Drug design and discovery process is costly and time consuming project, that too is led to increased time and cost burden due to later stage failure of drug design project. Many promising druggable candidates were failed in later stages and caused irreparable loss to the organization (Dibyajyoti et al., 2013).Thus to avoid a such problems one should be a careful for selection of molecule which is to be taken into next phase of drug development project (Kiriiri et al., 2020).The more credible experimental data make us sure about the druggability of any molecule but doing such traditional approach is costly and time consuming. However, computational approaches mimicking the experimental tools can be considered as versatile and powerful tool and can be used to generate the in-silico data. Different techniques such as HTVS, MD simulations studies, ADME calculation and molecular modelling approaches have been used for identification of HITs in the earlier stages of drug development phase (Mandal et al., 2009).The current study also considered the use of computational tool for identification of HITs against the COV-19 main protease and can be used for the treatment of deadliest ever pandemic by corona virus. The primary screening of the small database of phenolic compounds was performed by molecular docking studies. At the preliminary screening we identified top 10 HITs (Table 1) which were subjected to further studies for generation of in-silico data. Among these HIT-5 and HIT-6 formed the five & seven polar H bond withThr26; Gln189; Glu166; Asn142; Gly143 & Leu27; Tyr54; Val42; Pro52; Cys44; Met49; Met165 active site residues respectively indicating the formation of stable ligand–protein complex which could modulate the conformational changes incovid-19 main protease (Fig. 3). The presence of hydrophobic and polar interactions would lead to stabilization of complex (Moy et al., 1994). Hence, these two molecules were also probable promising candidate which we can take for further investigation. However, depending upon the docking score, comparative & identical interactions with Ebselen, the HIT-1 and HIT-2 was further subjected to detailed molecular simulations studies (Wang et al., 2020). The reason for generating the more in-silico data is the less credibility of initial docking results. Numerous reports have been published or many drug discovery projects have been failed due to false positive molecular docking results. Hence, we have decided to take top two identified HITs i.e. HIT-1 and HIT-2 for in depth molecular modelling studies. To know the stability of drug-ligand docked complex the top molecules were subjected to molecular dynamics simulation studies. The RMSD and RMSF values were calculated to see deviation of molecule from the active site. The obtained lower RMSD and RMSF values (Fig. 4) suggested the drug-ligand complexes were stable and molecules did not fluctuate from the active site protein. The different interactions present in docked complex (Fig. 3) were found to be retained over entire 50 ns run of MD simulations (Fig. 5) (Singh et al., 2022). Many times, the molecular docking studies gave the false positive results regarding binding affinities between ligand and protein complex. The reason behind this is lesser use of flexibility. Hence the more validated Poisson–Boltzmann surface area (MM/GBSA) based molecular mechanics calculation was done to determine the binding free energies of ligand–protein complexes. The sum of all interactions present between the ligand and protein is expressed in terms of binding free energies (−ΔG) (Sun et al., 2014). The obtained results were summarized in Table 2. The identical values of reference molecule and identified HITs suggested that the HITs could cause the conformational changes in covid-19 main protease as that of reference molecule and can produce the inhibitory effect. Thus, by using molecular docking, MD simulations, MMGB/SA binding free energies we could assume that the identified HITs would be the promising inhibitors ofcovid-19 main protease enzyme. This pharmacodynamics related in-silico data seems to be more creditable but there is always a worry about pharmacokinetic profiling of any newer molecule. To check and confirm the drug likeliness of identified molecules the reference and identified HITs were subjected to in-silico ADME calculations and the results obtained were displayed in Table 3. None of the molecules violate the Ro5 and Ro3 indicating the molecules had the drug likeliness properties except HIT1 (Sarkar et al., 2021). The other parameters of toxicity profiling were also found to be optimal and fall under the drug-like properties of the molecule. Further the identified molecules were subjected to in-silico CYPs metabolic studies. The major CYPs isoform responsible for xenobiotic metabolism CYP3A4, CYP2D6 & CYP2C9 were considered and specific site where the metabolism could be possible was determined. The obtained results were depicted in Fig. 6. The identification of potential metabolic sites in identified HITs would be helpful for further optimization and derivatization of drug like molecules (Rodrigues, 1994). The scores suggested that these molecules can absorb orally, well distributed in body and had the all druggable like properties. Hence, could be taken into further stages of drug design and discovery processes.
5 Conclusion
Structure based drug design approach was used to identify the potential drug candidate against the covid-19 infection. By considering the therapeutic importance and crucial role of protease enzyme in viral life cycle, covid-19 main protease (3Clpro) was used as a target. The small library of naturally occurring phenolic compounds was screened against the enzyme. Initially the molecular docking was done and identified top HITs were analysed and top scoring two molecules were further subjected to molecular dynamics simulations for better understanding of drug protein interaction even at atomic level. Furthermore, the binding free energy was also calculated to get the deeper insight into the ligand protein complex. The drug likeliness properties and druggability of molecules were also predicted by using the in-silico ADME calculations. The results were analysed and based upon the score obtained we identified two molecules HIT-1 (Corilagin) and HIT-2 (Oxyresveratrol) as the potential druggable candidates against thecovid-19 main protease. Based upon their comparative docking score, RMSD, RMSF values, binding free energies and in-silico ADME calculations we can say that the identified HITs could be the more promising candidate and can be taken into further stages of drug design and discovery processes and can be used for treatment and management of recent covid-19 infections.
6 Availability of data
Data will be available on request to corresponding.
7 Authors' contributions
Md Saquib Hasnainand Mohammed Aleissa were using Desmond V3 for design Structure-based drug design and virtual screening of a small library of phenolic compounds by using Desmond V3 for performing the molecular dynamics simulation from the scientific collaboratory protein data bank. Mohammed AL-Zharani and Saad Alkahtani were assessed the Molecular docking studies and Receptor grid generation. Md Saquib Hasnain was performed the MM-GBSA screening. Saad Alkahtani and Mohammed AL-Zharani were evaluated In-silico ADME prediction by using QikProp v3 to predict ADME properties. Mohammed Aleissa, Md Saquib Hasnain, Saad Alkahtani and Mohammed AL-Zharani were performed the statistical analysis and involved in the conception and design of the study.
Acknowledgments
This research was supported by the Deanship of Scientific Research, Imam Mohammad Ibn Saud Islamic University, Saudi Arabia, Grant No. (017-18-12-20).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- The risk factors associated with MERS-CoV patient fatality: a global survey. Diagn. Microbiol. Infect. Dis.. 2020;96(3):114876
- [Google Scholar]
- Protease targeted COVID-19 drug discovery: What we have learned from the past SARS-CoV inhibitors? Eur. J. Med. Chem.. 2021;215
- [Google Scholar]
- Bergdorf, M., Baxter S., Rendleman C. A., Shaw D. E. (2015). Desmond/GPU Performance as of October 2015. DE Shaw research.
- Screening a library of FDA-approved and bioactive compounds for antiviral activity against SARS-CoV-2. ACS Infect. Dis.. 2021;7(8):2337-2351.
- [Google Scholar]
- SARS-CoV-2 causing pneumonia-associated respiratory disorder (COVID-19): diagnostic and proposed therapeutic options. Eur. Rev. Med. Pharmacol. Sci.. 2020;24(7):4016-4026.
- [Google Scholar]
- Extensive partnership, collaboration, and teamwork is required to stop the COVID-19 outbreak. Arch. Med. Res.. 2020;51(7):728-730.
- [Google Scholar]
- Bioinformatics: The effects on the cost of drug discovery. Galle Med. J.. 2013;18(1):44.
- [Google Scholar]
- Role of proteolytic enzymes in the COVID-19 infection and promising therapeutic approaches. Biochem. Pharmacol.. 2020;182:114225.
- [Google Scholar]
- From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J.. 2014;281(18):4085-4096.
- [Google Scholar]
- An overview of safety assessment of the medicines currently used in the treatment of COVID-19 disease. Food Chem. Toxicol. 2020:111639.
- [Google Scholar]
- Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582(7811):289-293.
- [Google Scholar]
- Rational approach toward COVID-19 main protease inhibitors via molecular docking, molecular dynamics simulation and free energy calculation. Sci. Rep.. 2020;10(1):1-14.
- [Google Scholar]
- Exploring different approaches to improve the success of drug discovery and development projects: a review. Future J. Pharm. Sci.. 2020;6(1):1-12.
- [Google Scholar]
- Prolonged virus shedding even after seroconversion in a patient with COVID-19. J. Infect.. 2020;81(2):318-356.
- [Google Scholar]
- Liu, X., Zhang B., Jin Z., Yang H., Rao Z. 2020. The crystal structure of COVID-19 main protease in complex with an inhibitor N3. Protein DataBank 10.
- Coronavirus: Covid-19 Has Killed More People than SARS and MERS Combined, Despite Lower Case Fatality Rate. British Medical Journal Publishing Group; 2020.
- Computational studies on imidazo [1, 2-a] pyridine-3-carboxamide analogues as antimycobacterial agents: Common pharmacophore generation, atom-based 3D-QSAR, molecular dynamics simulation, QikProp, molecular docking and prime MMGBSA approaches. Open Pharm. Sci. J.. 2018;5(1):12-23.
- [Google Scholar]
- Intermolecular forces and energies between ligands and receptors. Science. 1994;266(5183):257-259.
- [Google Scholar]
- Organization, W. H. 2019. Middle East respiratory syndrome coronavirus (MERS-CoV).
- Use of in vitro human metabolism studies in drug development: an industrial perspective. Biochem. Pharmacol.. 1994;48(12):2147-2156.
- [Google Scholar]
- Targeting novel structural and functional features of coronavirus protease nsp5 (3CLpro, Mpro) in the age of COVID-19. J. Gen. Virol.. 2021;102(3)
- [Google Scholar]
- Schrodinger Software Suite. New York: Schrödinger; 2011.
- Comparative shape and electrostatic study of highly potent and selective CYP1B1 inhibitor: assessment of active site of CYP1B1 by binding mode analysis using site map tool. Indian J. Pharm. Educ. Res.. 2018;52(1):159-165.
- [Google Scholar]
- Comparative computational studies on selective cytochrome P450 1B1 inhibitors. Int. J. Bioautom.. 2020;24(3):213-224.
- [Google Scholar]
- In-silico evaluation of bioactive compounds from tea as potential SARS-CoV-2 nonstructural protein 16 inhibitors. J. Trad. Complement. Med.. 2022;12(1):35-43.
- [Google Scholar]
- A review of coronavirus disease-2019 (COVID-19) Indian J. Pediatr.. 2020;87(4):281-286.
- [Google Scholar]
- Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys. Chem. Chem. Phys.. 2014;16(40):22035-22045.
- [Google Scholar]
- Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: an observational cohort study. Lancet. Infect. Dis. 2020;20(5):565-574.
- [Google Scholar]
- Combined protein and ligand based physicochemical aspects of molecular recognition for the discovery of CDK9 inhibitor. Gene Reports. 2018;13:212-219.
- [Google Scholar]
- RS-WebPredictor: a server for predicting CYP-mediated sites of metabolism on drug-like molecules. Bioinformatics. 2013;29(4):497-498.
- [Google Scholar]
- Advances in the relationship between coronavirus infection and cardiovascular diseases. Biomed. Pharmacother.. 2020;127:110230.
- [Google Scholar]
- Coronavirus disease 2019 (COVID-19): a clinical update. Front. Med.. 2020;14(2):126-135.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2022.102283.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




