

Translate this page into:
qRT-PCR for enterovirus detection: Conversion to ultrafast protocols
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Enterovirus group (EV) still causes significant morbidity with economic impact worldwide. Quantitative RT-PCR (qRT-PCR) technology offers many advantages over to conventional RT-PCR in terms of rapidity and specificity. The TaqMan hydrolysis probe technique and Syber Green I intercalating dye strategy are by then largely used. Several published protocols are applied routinely for EV detection in food and environmental analysis advanced in chemical strategies and thermal profiles, in order to reduce the response times. In this study an Ultra-Fast protocol to detect and quantify EV RNA genome was tested and the application prospective of the protocol was described and discussed. The assay effectiveness was evaluated comparing two different set of primers/probe, targeting a 5′UTR region of EV genome. Three different oligonucleotides concentration were tested: 200 nM, 250 nM and 300 nM for TaqMan technique whereas 200 nM, 300 nM and 400 nM were employed for Syber Green I chemistry. The results demonstrated the validity of this Ultra-Fast approach, compared to the Traditional and Fast protocols. The best performance was obtained using 200 nM of proper oligonucleotides, in both of the chemical strategies tested. The response time of analysis was reduced at 50′ (probe) and 57′ (intercalating dye) per run, against the other longer protocols. The oligonucleotides features can affect the assay performance and should satisfy specific characteristics.
Keywords
Enterovirus
qRT-PCR
Ultra-Fast protocol

1 Introduction
Enterovirus group (EV) includes divers human enteric viruses, such as poliovirus, coxackievirus group A, coxackievirus group B, echovirus, etc, that still causes significant morbidity with economic impact in both developed and not developed countries (Dubot-Pérès et al., 2014; Hindiyeh et al., 2014; Zhang et al., 2014). EV persists in the environment for long time especially in the water bodies and may be transfers in food, keeping the chain of infection. In environmental matrices the EV load is variable, very low in the waters bodies compared to the food and/or the wastewater matrices. For this reason, a lot of time is consumed for the pretreatment and the concentration steps needful prior to evaluate the presence of viral particles.
Active environmental surveillance protocols require sensitive and specific tests which may reduce times and manipulation of samples at least during the detection assays respect to the time consuming culture isolation method. Molecular tests as the latest quantitative Reverse-Transcription Polymerase Chain Reaction (qRT-PCR) represent an excellent and rapid analytical tool giving accuracy, efficiency and reproducibility of the assays (Bustin, 2005). Among fluorogenic chemical strategies the TaqMan hydrolysis probe and the Syber Green I intercalating dye techniques are commonly used in qRT-PCR and several published protocols have entered into the routine for EV detection in food and environmental matrices (Nijhuis et al., 2002; Donia et al., 2005, 2010; Dubot- Pérès et al., 2014; Jebri et al., 2014). The amplification of the highly conserved 5′-untraslated region (5′UTR) of EV genome is still largely used in environmental surveillance and clinical analyses (Kurdziel et al., 2001; Donaldson et al., 2002; Donia et al., 2010; Gervasi et al., 2012; Harwood et al., 2013; Zhang et al., 2014; Jebri et al., 2014; Kaas et al., 2016) because can detect all members of the EV group (Yates, 2014).
A qRT-PCR protocol for EV detection has been optimized by Donia et al. (2005) and applied in environmental assays (Donia et al., 2010; Masciopinto et al., 2011). Advancing in biochemical strategies and thermal profiles have been gradually enhanced the published protocols, reducing analysis response times. For this purpose the industries have improved the instruments and the performances of the reagent mixes suggesting Ultra-Fast run formulations, to ensure reliable and reproducible data in shorter times by the qRT-PCR application. Indeed, during the years, the qRT-PCR run times were reduced, on average, from 220 min to 120 min per test consequent the technology enhancements. The latest generation instruments, updated by new spectra optical systems and analysis software, promise superior precision data in 50 min per test using Ultra-Fast chemistry approaches. By the protocols conversion it is expected energy and cost savings, over the time, without to affect data analysis. Based on the current knowledge there are no data in the literature about the application of Ultra-Fast protocols for enteric virus’s detection.
In this paper the results of this application were exposed and preliminary results were discussed in order to consider this procedure in environmental viruses monitoring. For this purpose two qRT-PCR protocols were compared: the first one optimized by Donia et al. (2005) and successfully employed by researchers in environmental (Le Guyader et al., 2008; Vilariño et al., 2009) and clinical assays (Le Guyader et al., 2008), the second suggested by Donaldson et al. (2002) and widely cited in several articles and used to date by researchers in the environmental EV surveillance and food safety (Tuladhar et al., 2012; Zhang and Wang, 2014; Kaas et al., 2016).
2 Materials and method
2.1 Experimental design
A comparison between two qRT-PCR protocols in use for EV detection and quantification was developed converting the thermal profile in Ultra-Fast run.
The protocols conversion was evaluated applying the chemical strategies commonly used TaqMan hydrolysis probe and Syber Green I intercalating dye, testing different oligonucleotides concentration.
2.2 Stool sample processing and positive controls preparation
To assess the applicability of the protocol a stool sample positive for EV genome presence was processed as unknown specimen. Stool sample was acquired from hospitalized children, diagnosed with acute gastroenteritis and screened for the panel of enteric viruses by RT-PCR end point. Feces were diluted with phosphate-buffered saline (PBS) (Merck) or 0.89% NaCl to 10% suspensions. After vigorous mixing the fecal specimen was clarified by centrifugation at 2,500g for 20 min at 4 °C, and then ultracentrifuged to remove any molecular test inhibitors in a bench Beckman ultracentrifuge Optima TL (Milan, Italy) equipped with a TLA-100 rotor at 50,000g for 1 h at 4 °C.
Three enteroviruses were chosen to simulate positive specimens and as positive EV controls: Poliovirus 1 Sabin type (P1S), Coxsackievirus B4 (CB4) and Echovirus 9 (Ecv9). Viruses were cultured as previously described (Donia et al., 2005) and titrated in laboratory by qRT-PCR.
Sample and controls were tested in two replicates each one. Extraction and amplification effectiveness was evaluated using the synthetic RNA standard cloned, tested as the reaction control at several known concentrations, 1 × 104, 1 × 102, 1 × 101 gce/µL.
RNA was extracted by the QIAmp viral RNA kit (Qiagen, Milan, Italy) following the manufacturer’s instructions and dissolved in RNase -free water containing 0, 5 U/μL of RNase inhibitor (Promega, Milan, Italy).
2.3 Standard cloning
A 263 bp fragment of the 5′UTR region of the complete sequence of P1S strain (Accession number AY184219, position 428–690 nucleotides) was cloned in pCR 4 TOPO vector (Invitrogen- Milan, Italy), in accordance with the manufacturer’s recommendations. Synthetic RNA was transcribed in vitro (T7 RNA polymerase – Promega, Milan, Italy) and after digested with Dnase and Rnase-free RQ1 (Promega, Milan, Italy). Synthetic RNA was purified with the QIAmp viral RNA kit (Qiagen, Milan, Italy) following the manufacturer’s instructions and dissolved in RNase -free water containing 0, 5 U/μL of RNase inhibitor (Promega, Milan, Italy). The synthetic RNA yield was determined spectrophotometrically (NanoDrop- Euroclone - Milan, Italy) and converted in gce/μL (1 × 108 gce/μL, stock solution).
2.4 Primers/probe design
The primers/probe set (Ts/Tas/Tp) was designed as previously described (Donia et al., 2005) according to the guide lines for qRT-PCR performance (Beacon Designer-PREMIER Biosoft-online tool for qPCR assays and by Oligo Evaluator-SIGMA-ALDRICH). Comparison was carry out versus the primers/probe set (Up/Dw/Pr) suggested by Donaldson et al. (2002) and recognizing the same genome region of the standard cloned. Oligosequences are reported in Table 3. #Traditional qRT-PCR protocol; *Fast qRT-PCR protocol; §Ultra-Fast qRT-PCR protocol; NA = No Amplification.
Ts/Tas/Tp (Donia et al., 2005)
Primers/probe concentration
Slope values (means)
CC (R2) (means)
EFF (means)
TaqMan 220 min.#
200 nM
−3.3
0.991
99.5%
250 nM
−3.0
0.886
109%
300 nM
−4.0
0.966
76%
TaqMan 120 min.*
200 nM
−3.4
0.999
98%
250 nM
−3.0
0.994
106%
300 nM
−2.9
0.975
114%
TaqMan 50 min.§
200 nM
−3.2
0.998
102%
250 nM
−2.2
0.960
134%
300 nM
−2.0
0.890
140%
SybGr I 240 min.#
200 nM
−3.9
0.995
78%
300 nM
−3.7
0.996
85%
400 nM
−3.3
0.997
100%
SybGr I 150 min.*
200 nM
−3.2
0.998
99%
300 nM
−3.0
0.995
97%
400 nM
−2.8
0.988
106%
SybGr I 57 min.§
200 nM
−3.2
0.999
102%
300 nM
−2.9
0.988
106%
400 nM
−2.5
0.899
141%
Up/Dw/Pr
Donaldson et al. (2002)
TaqMan 220 min.#
600/250 nM
−3.4
0.999
98%
400 nM
−2.4
0.898
150%
TaqMan 120 min.*
600/250 nM
NA
NA
NA
200 nM
NA
NA
NA
TaqMan 50 min.§
600/250 nM
NA
NA
NA
200 nM
NA
NA
NA
Thermal profiles¥
RT
Denaturation and activation Taq enzyme
Denaturation
Annealing
Extension
N° cycles
Traditional protocol
TaqMan
50 °C × 45’
95 °C × 10’
94 °C × 15’’
60 °C × 30’’
72 °C × 30’’
45
Syber Green I*
50 °C × 45’
95 °C × 10’
94 °C × 15’’
45 °C × 30’’
72 °C × 30’’
45
Fast protocol
TaqMan
55 °C × 30’
95 °C × 13’
94 °C × 15’’
60 °C × 1’
45
Syber Green I*
50 °C × 30’
95 °C × 13’
94 °C × 15’’
55 °C × 30’’
72 °C × 30’’
45
Ultra-Fast protocol
TaqMan
50 °C × 10’
95 °C × 3’
95 °C × 5’’
60 °C × 20’’
40
Syber Green I*
50 °C × 10’
95 °C × 3’
95 °C × 5’’
60 °C × 20’’
40
Oligonucleotide sequences
Secondary structures
Strength
Up*
GGCCCCTGAATGCGGCTAAT
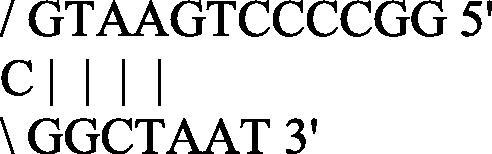
Moderate
Dw*
CACCGGATGGCCAATCCAA
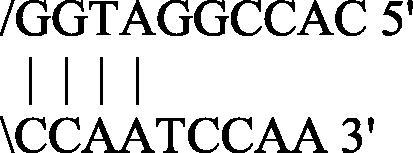
Strong
Pr*
CGGACACCCAAAGTAGTCGGTTCCG
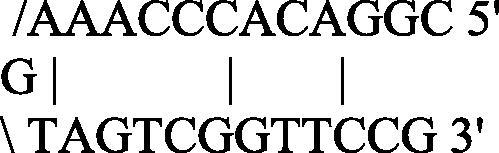
Strong
Ts§
GAATGCGGCTAATCCCAACCTC
None
Tas§
GGAAACACGGACACCCAAAGTAG
None
Tp§
TGCGCGTTACGACAGGCCAATCACT
None
Both probes (Tp-Pr) were labeled according to the TaqMan fluorogenic chemical strategy using FAM (6-carboxyfluorescein) at the 5′ end and TAMRA (6-carboxy-N, N, N0, and N0-tetramethylrhodamine) at the 3′end of the sequences.
2.5 Calibration curve
The calibration curve was generated as previously described (Donia et al., 2005) by performing 10-fold dilutions series of the synthetic RNA standard using five replicates per run.
2.6 qRT-PCR (one-step)
Traditional qRT-PCR protocols (∼220 min or more) were performed in the Bio Rad iCycler iQ instrument (Milan-Italy) while Fast protocols (120–150 min) were carried out with the Mastercycler ep realplex instrument (Eppendorf – Germany); in both of cases the Qiagen master mixes were employed for the assays (QuantiTect Probe RT-PCR kit and QuantiTect Syber Green I RT-PCR kit-Hilden, Germany). Ultra-Fast protocols were run in the AriaMx Realtime System (Agilent Technologies-United States) using Brillant III Ultra-Fast qRT-PCR and Brillant III Ultra-Fast SYBER Green qRT-PCR master mixes (Stratagene- Agilent Technologies-United States). Optimized protocols suggested by the manufacturers were adjusted for each assay to obtain the best performance. In particular, in the final volume of 20 µL three different concentrations of two primers/probes sets were tested: 200 nM, 250 nM and 300 nM. Syber Green I chemical strategy was evaluated using 200 nM, 300 nM and 400 nM of primers concentration.
Five replicates of the standard dilutions and two replicates for controls were amplified in each run. Analyses were confirmed by adding a stool sample positive for EV RNA genome presence (two replicate × run). Before tests, the sequences of primers/probe set suggested by Donaldson et al. (2002) were aligned with the cloned standard sequence to verify the 5′UTR fragment position (MultAlin software Corpet, 1988). Protocols comparison was carried out using the reaction conditions reported by Donaldson et al. (2002) in Fast and Ultra- Fast thermal profiles combining primers/probe concentrations. All assays were repeated at least three times.
2.7 Thermal profiles
Thermal profile conditions evaluated in this study were reported in Table 2. The differences among the profiles, concerning the cycle’s number, the running times and the temperatures applied, were itemized.
3 Results
Ultra-Fast qRT-PCR protocol was tested in order to evaluate the applicability of this approach for the detection and quantification of EV RNA genome. A comparison among protocols just in use was performed. Data analysis of the evaluation assays were shown in Table 1.
According to the mathematic basic equation model for quantitative analysis, a very good amplification efficiency was obtained both TaqMan hydrolysis probe and Syber Green I intercalating dye chemistry using Ultra-Fast Thermal profiles: slope values −3.2 on average, ≈102% EFF. Correlation Coefficient (R2) values represented the reproducibility of the assays, on average of 0,998 and 0,999 respectively. The best result was achieved using 200 nM of Ts/Tas/Tp primers/probe and Ts/Tas primers concentration respectively. The EV RNA genome was amplified in the stool positive sample and quantified in 1, 23 × 106 gce/μL. Three positive control viruses (P1S, CB4, Ecv 9) were included in amplification run (3 × 105 gce/μL, 1 × 103 gce/μL, 2 × 103 gce/μL respectively) and each one was confirmed in the amount. Equally, the synthetic RNA known concentrations tested as reaction controls were confirmed in all of runs performed, demonstrating a low RNA amount detection in the quantification assay (1 × 101 gce/μL), and respect to the standard dilution input. 200 nM of oligonucleotides concentration was also tested using Traditional and Fast qRT-PCR thermal profiles. The results showed very good amplification efficiency: slope values −3.3 ≈ 99, 5% EFF and −3.4 ≈ 98% EFF respectively, by using TaqMan chemistry and the protocol conditions assay reported in Table 2. The Syber Green I intercalating dye chemical strategy allowed the best performance applying Fast and Ultra-Fast thermal profiles: slope values −3.2 ≈ 99% EFF; slope values −3.2 ≈ 102% EFF respectively.
A very low sensitivity and specificity in the assay were exhibited applying the Ultra-Fast thermal profile and by using 250 nM and 300 nM of the primers/probe concentrations: slope values −2.2 and −2.0 respectively. Syber Green I intercalating dye chemistry displayed similar results against the improved values showed by Traditional thermal and protocol conditions (Table 1).
The protocol effectiveness as well as the evaluation of the assays performance were verify applying the primers/probe set proposed by Donaldson et al. (2002) in Fast and Ultra-Fast qRT-PCR conditions. No standard curve was generated by the dilutions standard due to the failure of nucleic acid amplification, as well as for positive controls and the stool positive sample added in each run. Tests were repeated several times without changing the conditions applied and described both in the thermal profile and the primers/probe concentrations. The results were equivalent to those obtained before. Applying the Traditional thermal profile and protocol conditions described by Donaldson et al. (2002) qRT-PCR assay was comparable in performances, after longer run times (Table 1).
4 Discussion
A comparison of qRT-PCR protocols was performed to detect and quantify EV genome in a short time (∼50 min–57 min). Proper one-step-reaction master mixes and different oligonucleotides concentrations according to the previous protocols (Donaldson et al., 2002; Donia et al., 2005) were employed. Data acquired suggest some observations about the not working of the oligonucleotides proposed by Donaldson et al. (2002) in Ultra-Fast conditions. Various factors might be hypothesized that in this context will be presented and discussed.
The best concentration of primers and probe to use in qRT-PCR is not always of equal molarity but adjusted in accordance with the protocol researcher’s requirements. Conversely the design of oligonucleotides should be to comply at specific characteristics recommended by guidelines for primer design (Rychlik, 1993).
The presence of secondary structures in the oligonucleotides sequence should be avoid to perform qRT-PCR, as well as the complementary interplay among oligonucleotide sequences that are responsible of the primer dimers formation. Primers/probe previously designed (Donia et al., 2005) and used in this comparison study were screened carefully to highlight potential interferences with the qRT-PCR performance.
Oligonucleotides sequences of the Donaldson et al. (2002) protocol were examined in detail to understand the amplification failure. Algorithmic properties of primers and probes were reported in Table 3 and in Fig. 1.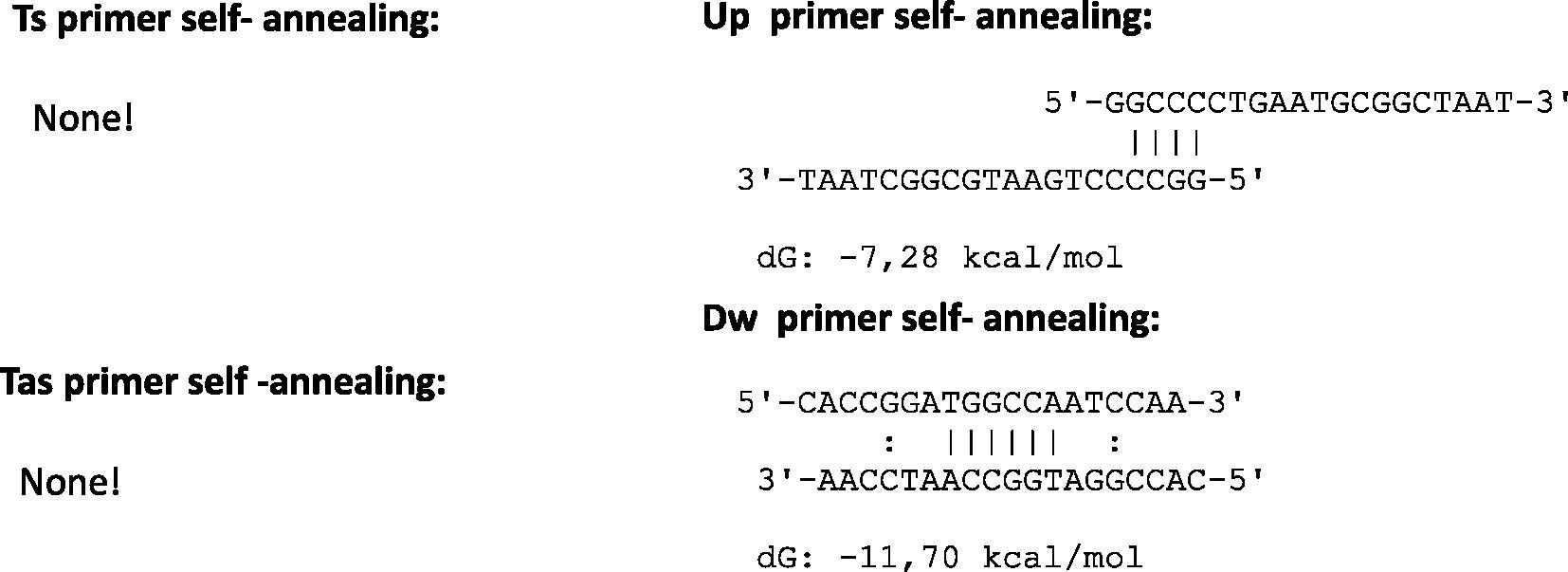
Compatibility of primers pairs compared in the study.
Algorithms were established under specific basic criteria: the melting temperature of the primers and the free energy (ΔG) of the secondary structures, if any, were calculated by the nearest neighbor method (Breslauer et al., 1986). The formation of a single stranded loop with a large negative ΔG value is an index of the structure’s stability (loop) typically unfavorable for the primer activation. Traditional thermal profiles provide a complete oligonucleotides linearization for the annealing/extension step in more minutes and at high temperature to allow specific amplicons. Primers with ΔG in hairpin loop approaching −3 Kcal/mol (at 25 °C) are troublesome when their 3′-end is “tied up”, because this can cause internal primer extension removing a given primer from the reaction (Rychlik, 1993). This condition requires a higher primers concentration molarity in the reaction mix to ensure a better yield of amplicons and prior a full amount of cDNA from the revers transcription step. Primes/probe set chosen by Donaldson et al. (2002) shows secondary structures with algorithm features just described. In particular, the Dw primer shows large negative ΔG values in self-annealing status.
It is reasonable to think that the use of proper designed oligonucleotides (Donia et al., 2005) has allowed a concentration reduction for the amplification (200 nM), as well as the time required for the protocol run (50 min). The optimized enzymes and other constituents in master mixes allow a promptly use of the oligonucleotides for the biomolecular reaction, having few seconds for a good performance in Ultra-Fast thermal conditions. The right oligonucleotides molarity, the temperature and the time needed to the annealing/extension step are closely linked to the secondary structures eventually present in the primers and probes sequences. The protocol proposed by Donaldson et al. (2002) employs a high concentration of upstream and downstream primers (600 nM) and a longer annealing/extension time (1 min.), typical of Traditional protocols, to permit the amplicons elongation.
The protocol described by Donia et al. (2005) has been optimized to obtain very good performances according to linear equation of the standard curve generated. The goal has been reached applying both Traditional that Fast qRT-PCR thermal profile by using a first and second generations of the Thermal Cycler unit (Le Guyader et al., 2008; Vilariño et al., 2009; Donia et al., 2010; Masciopinto et al., 2011). The set of primers/probe was selected intending to customize a protocol for environmental virology analyses. During the years this protocol was modified reducing the run times but without compromising the accuracy of data (Donia et al., 2010; Masciopinto et al., 2011). Lastly the protocol was converted in Ultra-Fast chemical and thermal profile to test the reliability of this approach in order to update the EV detection and quantification procedures.
To improving the performance of qRT-PCR an important role is doubtless played by the enzymes engineered for specific master mixes reaching superior levels of sensibility and specificity performed at high temperatures during the Ultra-Fast protocol. The reverse transcriptase (RT) enzymes provided in the latest generation kits are specifically formulated to transcript the total RNA in just 10 min. The reverse transcription temperature of 50 °C–55 °C improves the cDNA yield inhibiting secondary structures formation and the mutated Taq DNA polymerase enzyme is suitable for faster replication by a “hot start” capability that reduces nonspecific products amplification. Denaturation steps can be performed in 2 sec (Ultra-Fast protocols) against 2 min of the Traditional thermal profile, improving the amplification specificity and keeping to a minimum the run protocol times. Concentrations of primers and probes are to be optimized varying the range of primers and probe according to the chemical strategy chosen.
5 Conclusion
qRT-PCR offers the advantage to attempt to molecular biology researches in one-step run with a minimal RNA samples handling. Technological improvements of protocols, instruments e chemical strategies determine a larger popularity of this approach for nucleic acid detection (Bustin et al., 2005; Watzinger et al., 2006).
In the first instance, innovative reagents combined to the innovative instruments have been addressed by the market to improve gene expression, allele discrimination, genotyping tests and more. Afterwards, clinical and environmental analyses have been benefited from these technical innovations. The proof of principle of the protocol evaluated and exposed in this paper seems to prove the Ultra-Fast protocol effectiveness in the EV RNA genome quantification. This represents an opportunity to halve the response time without penalize specificity and efficiency of qRT-PCR performance. The comparison carried out between the two protocols (Donia et al., 2005; Donaldson et al., 2002) seems to confirm this hypothesis.
Traditional and Fast qRT-PCR protocols, long since in use, remain valid and enforceable by the first and/or second generation of Thermal Cycler instruments not able to perform Ultra-Fast run. Nevertheless, each Traditional protocol used in qRT-PCR can be converted in Ultra-Fast protocol, employing specific instruments and special master mixes but following key criteria. Algorithmic of primers/probes should be verified to establish the presence of any secondary structures in the sequences, crucial for the success of Ultra-Fast protocols. On the other hand, after a few years thermal cycler instruments become obsolete and will be replaced with others more innovative. The new generation instruments anyway allow Traditional and Fast protocols runs within the required time; the opposite is not applicable. Detection and quantification of the EV genome is yet important in clinical specimens and environmental matrices. Viruses detection could have faster answer times since the samples pretreatment (concentration steps) is time consuming and cannot be still by-passed and/or reduced in the process, as well as the cell cultured methods to attempt to viruses identification. Ultra-Fast qRT-PCR protocol promises energy and cost savings that is advisable in environmental enteric viruses’ analyses.
The qRT-PCR using TaqMan hydrolysis probe technique is largely used but the Syber Green I intercalating dye chemistry offer an effective chance in some analysis contexts. EV RNA detection and quantification may be achieved in little less than one hour, both in clinical than in environmental analyses, by a simpler and faster method than conventional molecular approaches.
As mentioned just before, there are no data in the literature about the conversion of thermal cycles protocol that run in around 50 min for TaqMan qRT-PCR and 57 min (included the melting curve analysis) for Syber Green I qRT-PCR. The maximum accomplished by the other known Fast protocols is about 120 min.
Further analyses need to enrich the experience in this field; it is considering applying this approach for other enteric viruses’ detection.
References
- Predicting DNA duplex stability from the base sequence. Proc. Natl. Acad. Sci. U.S.A.. 1986;83:3746-3750.
- [Google Scholar]
- Bustin, S.A., 2005. Real-Time PCR. Encyclopedia of Diagnostic Genomics and Proteomics. Ed. Marcel Dekker, New York, pp. 1117–1125.
- Quantitative real-time RT-PCR- a perspective. J. Mol. Endocrinol.. 2005;34:597-601.
- [Google Scholar]
- Multiple sequence alignment with hierarchical clustering. Nucl. Acids Res.. 1988;16:10881-10890.
- [Google Scholar]
- Detection, quantitation and identification of enteroviruses from surface waters and sponge tissue from the Florida Keys using real-time RT–PCR. Water Res.. 2002;36:2505-2514.
- [Google Scholar]
- Use of armored RNA as a standard to construct a calibration curve for real-time RT-PCR. J. Virol. Methods. 2005;126:157-163.
- [Google Scholar]
- Statistical correlation between enterovirus genome copy numbers and infectious viral particles in wastewater samples. Lett. Appl. Microbiol.. 2010;50:237-240.
- [Google Scholar]
- SYBR Green Real-Time PCR for the detection of all Enterovirus-A71 genogroups. PLoS One. 2014;9:e89963.
- [Google Scholar]
- Viral invasion of the amniotic cavity (VIAC) in the midtrimester of pregnancy. J. Matern. Fetal Neonatal Med.. 2012;25:2002-2013.
- [Google Scholar]
- Performance of viruses and bacteriophages for fecal source determination in a multi-laboratory, comparative study. Water Res.. 2013;47:6929-6943.
- [Google Scholar]
- Development and validation of a real time quantitative reverse transcription polymerase chain reaction (qRT-PCR) assay for investigation of wild poliovirus type 1-South Asian (SOAS) strain reintroduced into Israel, 2013 to 2014. Euro Surveill.. 2014;19:20710.
- [Google Scholar]
- A comparison of two extraction methods for the detection of Enteroviruses in raw sludge. J. Virol. Methods. 2014;200:1-5.
- [Google Scholar]
- A 1-year study on the detection of human enteric viruses in new caledonia. Food Environ. Virol.. 2016;8:46-56.
- [Google Scholar]
- Survival of poliovirus on soft fruits and salad vegetables. J. Food Prot.. 2001;64:706-709.
- [Google Scholar]
- Aichi virus, norovirus, astrovirus, enterovirus, and rotavirus involved in clinical cases from a French oyster-related gastroenteritis outbreak. J. Clin. Microbiol.. 2008;46:4011-4017.
- [Google Scholar]
- Analytical solution for the modeling of the natural time-dependent reduction of waterborne viruses injected into fractured aquifers. Environ. Sci. Technol.. 2011;45:636-642.
- [Google Scholar]
- Rapid and sensitive routine detection of all members of the genus Enterovirus in different clinical specimens by real-time PCR. J. Clin. Microbiol.. 2002;40:3666-3670.
- [Google Scholar]
- Selection of primers for polymerase chain reaction. In: White B.A., ed. PCR Protocols. Current Methods and Applications. Totowa, New Jersey: Humana Press; 1993. p. :31-40. Chapter. 2
- [Google Scholar]
- Thermal stability of structurally different viruses with proven or potential relevance to food safety. J. Appl. Microbiol.. 2012;112(5):1050-1057.
- [Google Scholar]
- Assessment of human enteric viruses in cultured and wild bivalve molluscs. Intern. Microbiol.. 2009;12:145-151.
- [Google Scholar]
- Detection and monitoring of virus infections by real-time PCR. Mol. Aspects Med.. 2006;27:254-298.
- [Google Scholar]
- Enterovirus. In: Microbiology of Waterborne Diseases Microbiological Aspects and Risks. London: Editor: Elsevier Academic Press; 2014. p. :493-504. Chapter 25
- [Google Scholar]
- Distribution of enteric pathogens in wastewater secondary effluent and safety analysis for urban water reuse. Human Ecol. Risk Assess.. 2014;20:797-806.
- [Google Scholar]
- A one-step, triplex, real-time RT-PCR assay for the simultaneous detection of enterovirus 71, coxsackie A16 and pan-enterovirus in a single tube. PLoS One. 2014;9:e102724.
- [Google Scholar]




