

Translate this page into:
Plasmodium vivax HAP2 genetic diversity and population structure from worldwide clinical samples. A potential Transmission-Blocking malaria vaccine candidate
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background
Malaria infections due to Plasmodium vivax are increasingly becoming a significant public health problem and are directly impacting malaria elimination efforts in many countries. Recent functional studies have shown that a pre-fertilising antigen; gamete membrane fusion protein HAP2/Generative Cell specific 1 (GCS1) domain significantly reduces the formation of P. vivax oocysts in mosquitos, implying that it could be developed as a transmission-blocking vaccine.
Methods
In this study, the genetic diversity, natural selection, haplotype network analysis and population structure of PvHAP2/GCS1 full-length gene using worldwide samples from 16 countries are determined and the implications for its use as a potential vaccine candidate against P. vivax are discussed.
Results
The study revealed low levels of nucleotide diversity (π ∼ 0.003, SNPs = 95) and purifying selection for the full-length PvHAP2 gene indicating functional constraints. Among the 95 SNPs (65 synonymous and 30 non-synonymous), 15 non-synonymous substitutions were localised to the C-terminal domain of the protein (674a.a − 862a.a) majority of these non-synonymous substitutions were from two countries of Central Africa i.e. Cameroon and Gabon. Phylogeographic haplotype network analysis indicated that a major haplotype (Hap_2, n = 34) is shared among countries from Asian countries and South American countries. Population structure analysis using a robust Bayesian algorithm indicated the existence of two sub-populations of PvHAP2/GCS1 among the worldwide samples.
Conclusion
This study suggests that, due to low genetic diversity across 15 countries, and shared haplotype identified, the HAP2 gene can be a good target for vaccine development as a transmission-blocking vaccine with the potential to provide strain-transcending immunity. The C-terminal region from two countries from Central Africa (Cameroon and Gabon) identified high number of non-synonymous substitutions, which may indicate new adaption of parasites from these countries. However, more detailed field based studies with a larger sample size from these countries are needed.
Keywords
Plasmodium vivax
Transmission-blocking vaccine
Polymorphisms
Vaccines
hap2/GCS1

1 Introduction
In 2020, an estimated 241 million malaria cases were reported worldwide, with 6.2 million deaths (World Health Organisation 2022). Plasmodium vivax is a significant public health problem, disproportionately impacting the poorest and most vulnerable populations. The overall proportion of cases has reduced from 8 % in 2000 to 2 % in 2020, according to the World Malaria Report (2022). Investments in malaria control and elimination programs have significantly reduced global malaria cases in recent decades. However, in areas where both P. falciparum and P. vivax coexist, P. vivax is increasingly becoming the primary cause of malaria (Battle et al., 2019). Even though P. vivax is widely distributed, there are presently no effective vaccinations available (Antonelli et al., 2020). Of all documented cases, 29–75 % of P. vivax malaria occurs occurs in the regions of the Americas, Southeast Asia, and the Eastern Mediterranean area (Poespoprodjo et al., 2009). The parasite has a complex, multi-stage life cycle, which makes developing effective vaccines for Plasmodium species difficult. For example, a wide and stage-specific protein repertoire is expressed during the asexual and sexual life cycle stages of P. vivax parasites, intermediate hosts, namely mosquitoes, and definitive hosts, which are humans (Florens et al., 2002; Le Roch et al., 2004). It is therefore necessary to develop innovative and integrated malaria prevention and control measures. A highly efficacious malaria vaccine is crucial to control and eventually eradicate malaria. Transmission-blocking vaccines (TBVs) that decreases the transmission of the parasite from humans to mosquitoes represent a significant step towards the objective of eliminating malaria. TBVs stimulate the production of antibodies that specifically target malaria antigens found in sexual stages, including gametocytes, gametes, zygotes, and ookinetes. These antibodies play a vital role in reducing the density of oocysts in the mosquito's midgut. In particular, transmission-blocking vaccinations (TBVs) are regarded as a critical element in efforts to control and eradicate malaria worldwide (PARASITE, 2002; Saul, 2007; Wu et al., 2015; Acquah et al., 2019). A TBV may be an effective technique for the eradication of P. vivax infections because of the early and continuous generation of gametocytes during infection. This is a crucial factor for the transmission of the parasites via mosquitoes. TBVs target antigens in the malaria parasite's sexual and early sporogonic phases, as well as midgut proteins in the mosquito vectors (Wu et al., 2015; Acquah et al., 2019). Proteins which are involved in fertilization are identified as targets for the development of TBVs and several of these have been well characterised in P. falciparum which shows transmission-reducing activity and these include P48/45, P47, P25, P28, P230, AgAPN1 and gamete membrane fusion protein HAP2/Generative Cell specific 1 (GCS1) (Duffy and Kaslow, 1997; van Dijk et al., 2001; Dinglasan et al., 2007; Wu et al., 2008; Blagborough and Sinden, 2009; Marin-Mogollon et al., 2018). Among these, HAP2/c, P230 and P48/45 are pre-fertilization antigens, while P25 and P28 are post-fertilization antigens and AgAPN1 is a mosquito midgut antigen.
HAP2/GCS1 is categorized as a class II viral fusion protein and contains a cysteine-rich. domain. Initially, this protein was discovered in the plant species Arabidopsis thaliana and Lilium longiflorum (Mori et al., 2006; von Besser et al., 2006; Angrisano et al., 2017a). The HAP2/GCS1 domain is found in a wide variety of organisms, including Plasmodium species and in the green algae Chlamydomonas. In Plasmodium, HAP2/GCS1 protein is localized on the surface of male gametes (Mori et al., 2006; Liu et al., 2008). A conserved 'cd loop' sequence of the HAP2/GCS1 protein, which corresponds to amino acids (a.a.) 178–207 in P. falciparum and (a.a.) 174–205 in PbHAP2, is essential for membrane binding during fertilisation (Angrisano et al., 2017b; Fédry et al., 2017). Mouse antibodies against recombinant PfHAP2 expressed in a wheat germ cell-free system showed strong transmission-reducing activity (Miura et al., 2013). A recent study reported that anti-PvHAP2 antibodies substantially reduced the number of oocysts in mosquitoes suggesting that PvHap2 could be a potential TBV against P. vivax infections and aid in malaria elimination (Qiu et al., 2020). Altogether, these studies suggest that HAP2 is a potential transmission-blocking vaccine candidate however, very limited studies have been done to investigate genetic diversity studies from worldwide samples. A recent study from the Greater Mekong Subregion (GMS) conducted on a fragment from 13nt to 2574nt of the gene indicated limited genetic diversity among field samples within the region (Li et al., 2020a). Similarly, a study focusing only on the short GCSI domain i.e. 397nt – 2097nt of the HAP2 gene from Iran indicated limited genetic diversity too (Mehrizi et al., 2016). Therefore, in this study, the genetic diversity, natural selection, haplotype network analysis and population structure of PvHAP2/GCS1 full-length gene using worldwide samples from 16 countries are determined and the implications for its use as a potential vaccine candidate against P. vivax are discussed. Potential epitopes within different domains of the protein are identified too.
2 Materials and Methods
2.1 PvHAP2/GCS1 sequence data
Full-length PvHAP2 genes without having any ambiguous bases were retrieved from clinical samples from 16 countries along with two reference strains (Sal-1; PVX_094925 and P01; PVP01_0814300) from PlasmoDB, (https://plasmodb.org) database (Supplementary Table 1). The MegAlign, Lasergene v 7.0 (DNASTAR) software's CLUSTAL-W program was used to align all of the sequences. Using MEGA 5.0 software, phylogenetic and polymorphism analyses were performed. To determine the relationship between PvHAP2/GCS1 amino acid sequences and its ortholog species in P. falciparum (PF3D7_1014200), P. reichenowi (PRCDC_1013600), P. knowlesi (PKNH_0814100), P. ovale (PocGH01_08022600) and P. malariae (PmUG01_08030200) in interspecies level, phylogenetic analyses were carried out using deduced amino acid sequences and the Maximum Likelihood (ML) approach based on the Poisson correction model as outlined in MEGA 5.0. 1,000 bootstrap replicates were performed to test the robustness of the trees.
2.2 Sequence diversity and natural selection
The software DnaSP v5.10 was used to determine sequence diversity (π) (Librado and Rozas, 2009). DnaSP software was also used to assess the number of polymorphic sites, synonymous and non-synonymous substitution, number of singletons, parsimony informative sites, number of haplotypes (H), haplotype and nucleotide diversity in PvHAP2/GCS1. Graphical representation of nucleotide diversity was determined using a window length of 100 and a step size of 25 bp in the DnaSP software. Using Nei & Gojobori's approach (Nei and Gojobori, 1986) in MEGA 5.10 software, the rates of synonymous substitution per synonymous site (dN) and nonsynonymous substitution per nonsynonymous site (dS), which determine the natural selection, were calculated. Using MEGA 5.10 software's output of the deduced PvHAP2 amino acid sequence polymorphism, amino acid haplotypes were manually determined. Additionally, The Tajima's D, Fu & Li's D* and F* neutrality tests were performed in the DnaSP v5.10 software, to help determine natural selection. Tajima's D value is zero under neutrality. Tajima's D's negative value denotes population expansion, while its positive and significant value denotes positive selection/balancing. Using the DnaSP software, Fu and Li's D* and F* were also determined. A positive and any significant value of these testes suggest population contraction, while excess singletons and negative values suggest population expansion.
2.3 Haplotype network (Genealogical) analysis
The construction of haplotype networks is a commonly employed method for examining and graphically representing the connections between DNA sequences within a population or species. For haplotype network analysis was conducted considering all the 118 PvHAP2/GCS1 sequences using samples from 16 countries. NETWORK version 4.6.1.2 software was used to generate the PvHAP2/GCS1 gene haplotype network using the median-joining method (Fluxus Technology Ltd, Suffolk, UK).
2.4 Population structure of PvHAP2/GCS1
The STRUCTURE V2.3.4 software, which is based on a Bayesian Markov chain Monte Carlo (MCMC) model, was used to determine the population genetic structure of the P. vivax parasite population from 16 different countries (Earl and VonHoldt, 2012). The number of populations that are most likely to exist was calculated using an admixture model (K). One run was performed with K set to 1 to 10. For each run, 500,000 Markov chain Monte Carlo generations after a burn-in period of 50,000 steps were used. By analysing K values based on the rate of change in log probability (LnP(D)) between successive K-values and applying the STRUCTURE HARVESTER to the data, the most likely K-value was predicted (Earl and VonHoldt, 2012).
2.5 Epitope prediction
B cell epitopes are a type of antigenic determinant that can be utilised in the process of peptide-based vaccine development. These determinants can be portions of foreign proteins or antigens (Saha and Raghava, 2006). In this study, B cell epitopes were predicted in silico across the full-length PvHAP2 amino acid sequences in two independent prediction servers to increase the robustness of the analyses; the antibody epitope prediction server IEDB Analysis, by selecting the Emini Surface Accessibility Prediction model (Emini et al., 1985) and (Saha and Raghava, 2006). The Bcpred software uses amino acid properties, such as exposed surface, flexibility, polarity, and hydrophilicity, to make predictions about B cell epitopes. For a prediction to be made, the amino acid attributes must meet a threshold score of 2.38. The PvHAP2 amino acid sequence, which was shared among the majority of the countries (shared haplotype) was chosen for epitope prediction with both servers.
3 Results
3.1 PvHAP2/GCS1 sequence identity, polymorphisms, and phylogenetic relationship between p. Vivax HAP2/GCS1 and its ortholog species
Alignment and comparison of deduced amino acid sequences of P. vivax HAP2/GCS1 (Sal I strain) with its ortholog members in P. knowlesi, P. falciparum, P. reichenowi, P. ovale and P. malariae was found to be 85.7 %, 51.1 %, 51.1 %, 62.1 % and 64.9 % respectively. A schematic diagram of the full-length structure of the HAP2/GCS1 gene of P. vivax and 30 amino acid polymorphisms with their precise locations within each domain as observed within the 118 worldwide sequences are shown in (Fig. 1). The study revealed 18 PvHAP2 amino acid haplotypes from the 118 worldwide amino acid sequences, haplotype 3 (n = 35, 29.6 %) was shared among 11 countries (Columbia, Mexico, Peru, Thailand, India, China, Gabon, Madagascar, Myanmar, PNG, and Cambodia) (Figure S1). Other shared haplotype with lower number of samples included haplotype 1 (n = 25, 21 %), haplotype 4 (n = 24, 20 %) and haplotype 2 (n = 20, 16.9 %) (Figure S1). The phylogenetic analysis using deduced amino acid sequences indicated that PvHAP2/GCS1 is closer to its ortholog in the zoonotic malaria parasite P. knowlesi compared to its human infecting orthologs in P. falciparum, P. malariae and other primate malaria species i.e. P. coatneyi (Fig. 2).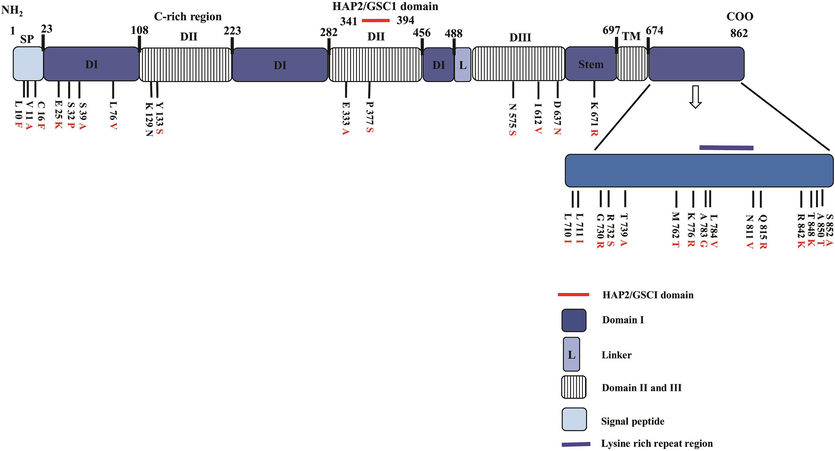
Domain organisation of PvHAP2 and amino acid polymorphism observed within 118 worldwide PvHAP2 sequences obtained from 16 countries. SP; Signal peptide, DI, DII, DIII, the domain I-III; L, linker (L); TM, transmembrane segment; C-rich region, the cysteine-rich region; PFAM10699, the HAP2/GSC1 domain.
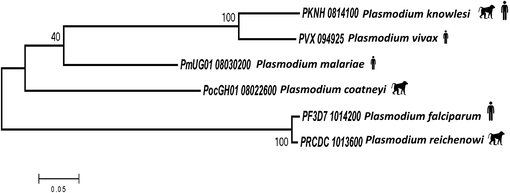
Phylogenetic relationship of PvHAP2 with its orthologs in Plasmodium knowlesi, Plasmodium malariae, Plasmodium coatneyi, Plasmodium falciparum and Plasmodium reichenowi. The silhouette figures of primate and humans indicates the host for each of the parasites.
3.2 PvHAP2 diversity and natural selection
DNA sequence analysis of 118 PvHap2 sequences revealed that there were 95 single nucleotide polymorphisms (SNPs) within 118 PvHAP2 sequences from 16 different nations, of which 65 were synonymous substitutions and 30 were non-synonymous substitutions (Figure S1). The hydrophobic “fusion loop” (cd loop) segment (167–198 a.a.) did not contain any non-synonymous substitution indicating conservation of membrane fusion function and gamete fertilisation. The overall nucleotide diversity (within all countries) was found to be π = 0.0030 which was slightly higher compared to a previous study done with samples only from the Greater Mekong Sub region (Li et al., 2020a) (Table 1). There were 68 parsimony informative sites within PvHAP2, 27 were singleton variable sites, and there were 25 haplotypes with a haplotype diversity of 0.842. (Table 1). The graphical representation of the nucleotide diversity and Tajima’ D values of PvHAP2/GCS1 is shown in (Fig. 3A) and (Fig. 3B) respectively. Thirty non-synonymous substitutions identified within the amino acid sequence are shown in (Figure S1).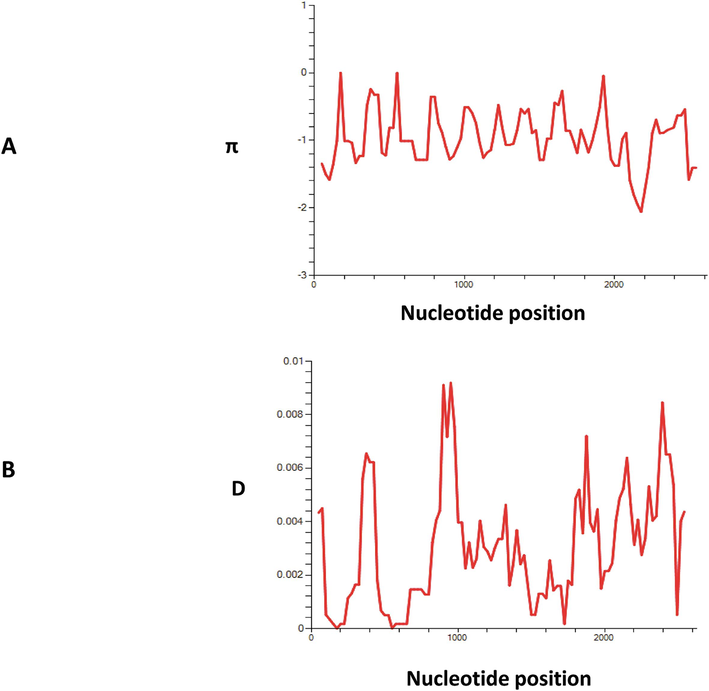
(A) Graphical representation of nucleotide diversity (π) within PvHAP2 gene and (B) Tajima’s D value. The window length and step size of π and D graph is 100 & 25 respectively as implemented in DnaSP software v5.0.
Natural selection analysis of PvHAP2/GCS1 gene from 118 sequences indicated the gene is under negative or purifying selection (dN-dS = -5.10, P > 0.1) probably due to functional constraints (Table 1). Overall, the Tajima’s D values were negative and significant (D = − 1.79, P < 0.05) indicating a strong purifying selection. The Fu & Li D* and F* values were also negative but not significant which indicates purifying selection and population expansion (Table 1).
Domain
No. of
Samples
SNPs
Syn
NonSyn
No. of
haplotypes
Diversity ± SD
Codon based
z-test dN─Ds
Fu & Li’s D*
Fu & Li’s F*
Taj D
Haplotype
Nucleotide
Full length
118
95
65
30
25
0.842 ± 0.018
0.0030 ± 0.00078
−5.1 (p > 0.1)
−1.78
(p > 0.10)−1.24 (p > 0.10)
−1.79
(p < 0.05)
3.3 Genealogical haplotype network analysis of PvHAP2/GCS1
The nucleic acid based haplotype network analysis of 25 haplotypes of the PvHAP2/GCS1 revealed that there were 4 major haplotype clusters (H_2, H_3, H_4 and H_9), of which, H_2, H_3 and H_4 formed a closely related group compared to Hap_9 indicating the presence of two major haplotype groups (Fig. 4). Among the major haplotypes, H_2 was shared within 11 countries and it had the highest number of samples (n = 34) (Fig. 4). The haplotype 9 (H_9) however, formed a separate cluster with maximum samples from Myanmar, China and Gabon. Minor shared haplotypes (H_7 and H_6) were observed within major haplotype clusters (H_2, H_3, H_4 and H_9).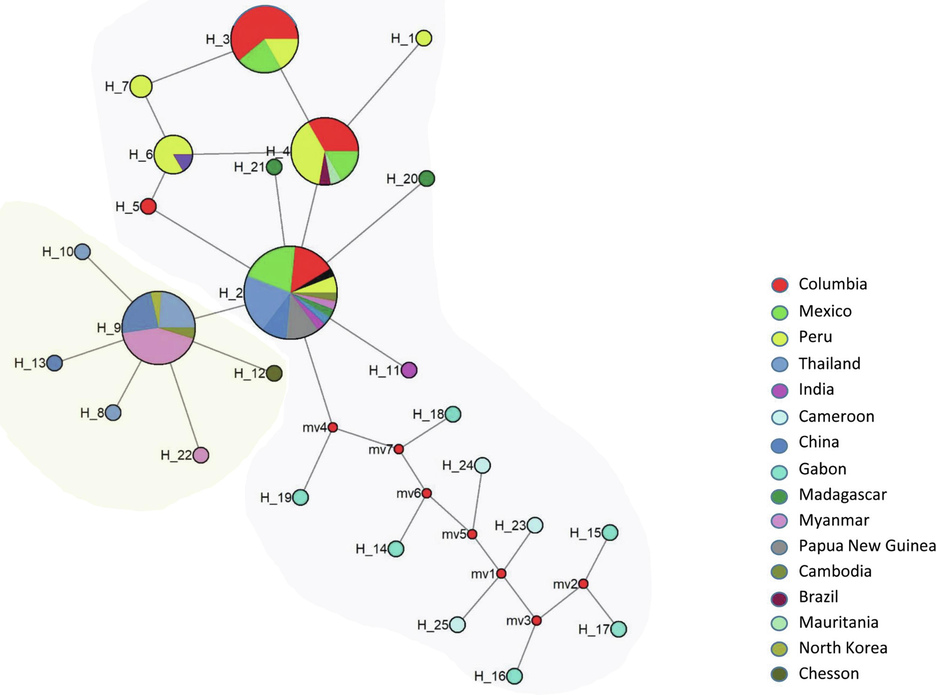
Haplotype network of PvHAP2 from 118 sequences from 16 countries. The highlighted clusters represents the two different subpopulation. Various colors represent different locations. Circles represent haplotypes with sizes proportional to haplotype frequency.
3.4 Population structure analysis of PvHAP2/GCS1 gene from 16 countries
A Bayesian algorithm-based population structure analysis of 118 PvHAP2/GCS1 sequences from 16 countries using STRUCTURE software revealed that there were two distinct sub-populations of the parasite within the worldwide samples (K = 2, ΔK = 503.64) (Fig. 5A and B).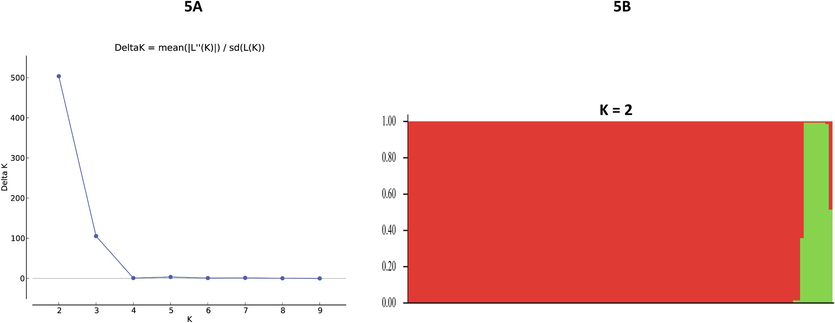
Most likely number of Plasmodium vivax parasite subpopulation clusters (K) within 16 countries based on Hap2 sequences (K = 2, ΔK = 503.64). Bayesian model–based STRUCTURE version 2.3.4 software was used.
3.5 B cell epitopes identified in PvHAP2
Based on amino acid haplotype analysis, the full-length PvHAP2 sequence of amino acid haplotype 3 was shared among 11 countries (FigureS1) for determining the B cell epitopes. Bcpred and IEDB servers identified 19 and 16 epitopes in P. vivax HAP2 amino acid sequence, respectively (Table 2) (Fig. 6). Epitope outputs from both servers were compared, and the results revealed that there are a total of 11 PvHAP2 epitopes, with their lengths varying from 5 to 26 peptides identified by both servers (Table 2). Among the common peptides identified, a lysine-rich repeat peptide was identified which had a high score from both the servers (782 KQTRRKKKKKKS 796).
Bcpred server
IEDB server
No.
Start AA
End AA
Peptide
Length
Start AA
End AA
Peptide
Length
1
1
5
MKPTR
5
21
27
RDKREDG
7
2
19
29
FGRDKREDGGR
11
117
131
HQKFKKYSEREIKEY
15
3
38
48
HSFAKKKVCTS
11
170
180
LFRDNKDIKRS
11
4
97
105
YLKYMKDIP
9
230
236
QEYSYDD
7
5
116
132
NHQKFKKYSEREIKEYT
17
249
256
TKEKKYEL
8
6
172
184
RDNKDIKRSKLKC
13
387
392
QRYYDA
6
7
246
258
DLVTKEKKYELND
13
401
406
GASKYK
6
8
402
410
ASKYKIKNV
9
413
425
SEPQTRIYKPSTL
13
9
415
422
PQTRIYKP
8
431
441
DKINNNHKKID
11
10
426
444
PDHLKDKINNNHKKIDAND
19
622
637
SVTKYDSEPEASQNDD
16
11
505
512
SNQSESKD
8
660
665
YYTKRI
6
12
537
543
TDKETGK
7
729
736
RGKRSKCK
8
13
575
581
NKEIKTM
7
774
780
RVKKSAK
7
14
587
598
RDSNGKECSKED
12
782
796
EALKQTRRKKKKKKS
15
15
624
631
TKYDSEPE
8
808
814
HSRNDYS
7
16
660
672
YYTKRIKNVIKKF
13
838
854
SQSDRSSNKGTRAPSRA
17
17
723
738
KERLHIRGKRSKCKKG
16
18
774
799
RVKKSAKSEALKQTRRKKKKKKSGRM
26
19
840
846
SDRSSNK
7
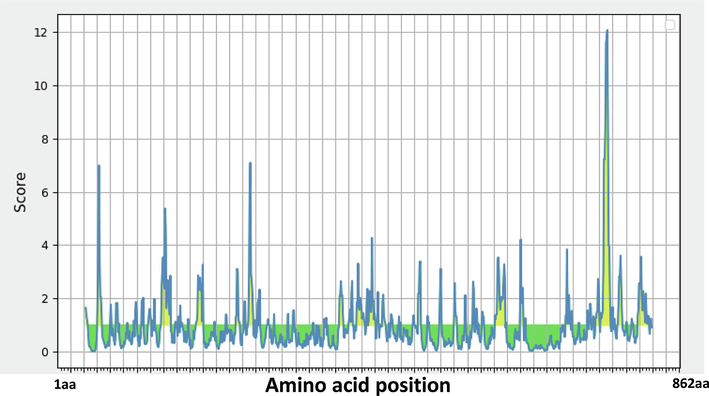
The graph represents the possible epitope region in PvHAP2 protein using epitope prediction software IEDB. The peaks (in yellow) in graph shows the possible epitope region.
4 Discussion and Conclusion
Although the World Health Organisation (WHO) considers that vaccines are one of the most effective strategies for the elimination and eradication of malaria, previous studies have shown that developing malarial vaccines is an extraordinarily difficult task. Two of the most significant obstacles in the process of designing a vaccine are allele-specific immune responses (Fluck et al., 2004) and the antigenic diversity of vaccine candidate antigens (Girard et al., 2007; Arnott et al., 2012). To design and develop a malaria vaccine that is effective on a global scale, it is vital to investigate the genetic diversity of vaccine candidate antigens in both low-endemic and high-endemic regions. The surface antigens of gametes and ookinetes are the primary focus of research and development for TBVs. In this study, the genetic diversity, phylogeography, population structure, and epitope characterization of Plasmodium vivax HAP2 full-length gene sequences were determined from worldwide clinical samples (n = 118, 16 countries) obtained from public databases. Previous population genetic studies have only focused on the GCSI domain (Mehrizi et al., 2016; Li et al., 2020a) of the HAP2 protein, which is essential for binding to host cells. Because of this, in the current study, the PvHAP2 full-length gene study from 16 countries was included. Amino acid sequence analysis indicated that the highest similarity was with the monkey malaria parasite P, knowlesi (∼85 %), this might positive implication towards development of cross-species vaccine candidates as reported in previous studies. Though 30 non-synonymous substitutions across the full-length protein were identified interestingly, 15 of these non-synonymous substitutions were localised towards the C-terminal region (674a.a. − 862a.a.) indicating immune selection pressure could be high in this domain. The localisation or accumulation of these non-synonymous substitutions in the C-terminal region could also indicate that this region might be exposed to host immune pressure. However, structural as well as functional studies along with field-based immunological studies need to be conducted to determine the immunogenicity of this domain. Previous studies have focused only on characterizing the HAP2 GCS1 domain of this protein (Qiu et al., 2020). Additionally, genetic diversity studies with a higher number of samples from each location would be essential too.
Analysis of PvHAP2 genetic diversity indices indicated lower levels of diversity (∼0.003) which was slightly higher than previously reported studies (Mehrizi et al., 2016; Li et al., 2020a). This may be due to the fact that this research concentrated primarily on the shorter segments rather than the full-length gene. Other TBVs such as Pvs28 and Pvs25 had shown similar levels (∼0.001––0.004) of genetic diversity in endemic areas of Myanmar and China (Feng et al., 2011; Le et al., 2019) indicating conserved protein function. It is interesting to note that despite low levels of nucleotide diversity, the haplotype diversity was found to be on the higher side for the PvHAP2 gene, indicating population expansion supported by negative values of Li and Fu’ D* and F* (−1.75 & −1.24). Natural selection analysis of worldwide PvHAP2 indicated purifying selection pressure acting on the whole gene which was not statistically significant but was similar to the previous studies and other TBVs (Feng et al., 2011; Mehrizi et al., 2016; Li et al., 2020b).
The haplotype network analysis indicated the presence of four major shared haplotypes within the 16 countries, which were grouped into two major groups. The robust Bayesian model-based population structure analysis also suggested that the PvHAP2 gene largely consisted of two sub-populations (K = 2, K = 503.6). This indicates that any vaccine design needs to take into consideration these two sub-populations to achieve high efficacy and worldwide coverage.
B cell epitope prediction using two different algorithms showed that 11 epitopes were predicted and among these a high scored C-terminal lysine-rich repeat region with 15 peptides EALKQTRRKKKKKKS could be a potential region where high immunogenicity could be found. The lysine-rich 15 amino acid peptide identified in this study could be potential TBV candidate for P. vivax, however, higher number of samples and functional studies would necessary to substantiate the presence of this epitope. This needs to be confirmed with functional as well as field-based immunological studies in the future. Moreover, it should be noted that only two countries, Gabon and Cameroon, both from Central Africa, out of the total 15 countries, have amino acid changes in the C-terminal region (as highlighted in the attached supplementary figure S1). This means that the majority of the field isolates function remained intact, except for parasite from these two countries. We believe that more field-based studies are needed from these countries to confirm whether natural selection is active. In addition, functional studies including sequences from these countries would be essential to understand the function, as we have noted that there are at least 8 amino acid changes in the C-terminal region, which could be important. This is because, the C-terminal region may prove to be crucial for vaccine and therapeutic strategy development, as previous studies on the functional analysis of the cytoplasmic domain of AMA1 have highlighted its significance in invasion and the development of novel therapeutic strategies (Treeck et al., 2009). The lysine-rich 15 amino acid peptide identified in this study could be potential TBV candidate for P. vivax, however, higher number of samples and functional studies would necessary to substantiate the presence of this epitope. Further confirmation through functional and field-based immunological studies on the C-terminal region will be necessary in the future.
5 Conclusion
This work is the first of its kind to disclose the genetic diversity as well as the population structure of the full-length PvHAP2 gene from 17 different nations. Although a low degree of genetic diversity and purifying selection were discovered, the C-terminal region indicated a higher number of non-synonymous substitutions. This finding suggests that this protein is vital to the survival and transmission of the parasite. The haplotype network and population structure analyses both pointed to the existence of at least two distinct subpopulations. This study uncovered a prevalent and shared PvHAP2 haplotype found all around the world, which is notable. In addition, the current investigation was able to identify probable B-cell epitopes over the entirety of the gene, as well as one high-scoring epitope towards the C-terminal region, which consisted of a lysine-rich 15 amino acid peptide. The findings of the current study will be very important in the rational design of a vaccine that can inhibit PvHAP2 transmission and has global application. However, functional studies focusing on immunogenicity and vaccine efficacy studies would be necessary.
Acknowledgement
The author extend his appreciation to the Deanship of Research and Graduate Studies at King Khalid University for funding this work through small group research under grant number RGP1/339/45.
References
- Transmission-Blocking Vaccines: Old Friends and New Prospects. Infect Immun. 2019;87
- [Google Scholar]
- Targeting the Conserved Fusion Loop of HAP2 Inhibits the Transmission of Plasmodium berghei and falciparum. Cell Rep. 2017;21:2868-2878.
- [Google Scholar]
- Understanding the population genetics of Plasmodium vivax is essential for malaria control and elimination. Malar J. 2012;11:14.
- [Google Scholar]
- Mapping the global endemicity and clinical burden of Plasmodium vivax, 2000–17: a spatial and temporal modelling study. Lancet. 2019;394:332-343.
- [Google Scholar]
- Plasmodium berghei HAP2 induces strong malaria transmission-blocking immunity in vivo and in vitro. Vaccine. 2009;27:5187-5194.
- [Google Scholar]
- Disruption of Plasmodium falciparum development by antibodies against a conserved mosquito midgut antigen. Proc Natl Acad Sci U S A. 2007;104:13461-13466.
- [Google Scholar]
- A novel malaria protein, Pfs28, and Pfs25 are genetically linked and synergistic as falciparum malaria transmission-blocking vaccines. Infect. Immun.. 1997;65:1109-1113.
- [Google Scholar]
- Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J Virol. 1985;55:836-839.
- [Google Scholar]
- The ancient gamete fusogen HAP2 is a eukaryotic class II fusion protein. Cell. 2017;168(904–915):e910.
- [Google Scholar]
- Genetic diversity of transmission-blocking vaccine candidates Pvs25 and Pvs28 in Plasmodium vivax isolates from Yunnan Province. China. Parasit Vectors. 2011;4:224.
- [Google Scholar]
- A proteomic view of the Plasmodium falciparum life cycle. Nature. 2002;419:520-526.
- [Google Scholar]
- Strain-specific humoral response to a polymorphic malaria vaccine. Infect Immun. 2004;72:6300-6305.
- [Google Scholar]
- A review of human vaccine research and development: malaria. Vaccine. 2007;25:1567-1580.
- [Google Scholar]
- Genetic diversity and natural selection of transmission-blocking vaccine candidate antigens Pvs25 and Pvs28 in Plasmodium vivax Myanmar isolates. Acta Trop. 2019;198:105104
- [Google Scholar]
- Global analysis of transcript and protein levels across the Plasmodium falciparum life cycle. Genome Res. 2004;14:2308-2318.
- [Google Scholar]
- Plasmodium vivax HAP2/GCS1 gene exhibits limited genetic diversity among parasite isolates from the Greater Mekong Subregion. Parasit. Vectors. 2020;13:1-11.
- [Google Scholar]
- Plasmodium vivax HAP2/GCS1 gene exhibits limited genetic diversity among parasite isolates from the Greater Mekong Subregion. Parasit Vectors. 2020;13:175.
- [Google Scholar]
- The conserved plant sterility gene HAP2 functions after attachment of fusogenic membranes in Chlamydomonas and Plasmodium gametes. Genes Dev.. 2008;22:1051-1068.
- [Google Scholar]
- The Plasmodium falciparum male gametocyte protein P230p, a paralog of P230, is vital for ookinete formation and mosquito transmission. Sci. Rep.. 2018;8:1-13.
- [Google Scholar]
- Worldwide population genetic analysis and natural selection in the Plasmodium vivax Generative Cell Specific 1 (PvGCS1) as a transmission-blocking vaccine candidate. Infect Genet Evol. 2016;43:50-57.
- [Google Scholar]
- Functional comparison of Plasmodium falciparum transmission-blocking vaccine candidates by the standard membrane-feeding assay. Infect Immun. 2013;81:4377-4382.
- [Google Scholar]
- GENERATIVE CELL SPECIFIC 1 is essential for angiosperm fertilization. Nat. Cell Biol.. 2006;8:64-71.
- [Google Scholar]
- Development of malaria vaccines that block transmission of parasites by mosquito vectors. J. Med. Invest.. 2002;49:119.
- [Google Scholar]
- Vivax malaria: a major cause of morbidity in early infancy. Clin Infect Dis. 2009;48:1704-1712.
- [Google Scholar]
- Evaluation of Plasmodium vivax HAP2 as a transmission-blocking vaccine candidate. Vaccine. 2020;38:2841-2848.
- [Google Scholar]
- Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins. 2006;65:40-48.
- [Google Scholar]
- Mosquito stage, transmission blocking vaccines for malaria. Curr. Opin. Infect. Dis.. 2007;20:476-481.
- [Google Scholar]
- A central role for P48/45 in malaria parasite male gamete fertility. Cell. 2001;104:153-164.
- [Google Scholar]
- Von Besser, K., Frank, A.C., Johnson, M.A., and Preuss, D. (2006). Arabidopsis HAP2 (GCS1) is a sperm-specific gene required for pollen tube guidance and fertilization.
- World Malaria Report, W.H.O. (2022).
- Phase 1 trial of malaria transmission blocking vaccine candidates Pfs25 and Pvs25 formulated with montanide ISA 51. PLoS One. 2008;3:e2636.
- [Google Scholar]
- Development of malaria transmission-blocking vaccines: from concept to product. Adv Parasitol. 2015;89:109-152.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2024.103269.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




