

Translate this page into:
Pharmacokinetics of some newly synthesized 1, 5- benzothiazepine scaffolds: A molecular docking and molecular dynamics simulation approach
⁎Corresponding authors. klameta77@hotmail.com (Keshav Lalit Ameta), klameta@spuvvn.edu (Keshav Lalit Ameta), shafiul.haque@hotmail.com (Shafiul Haque),
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
A highly efficient in silico pharmacokinetics was studied for some newly synthesized 1, 5- benzothiazepine derivatives. A total of fourteen title moieties were prepared by the condensation of substituted chalcones and 2- aminothiophenol that led to ring expansion in the presence of CuO nanocatalytic framework. The reported synthetic route is advantageous due to shorter reaction time and enhanced yield. The drug-target binding analysis revealed the potential therapeutic targets against 1,5- benzothiazepine derivatives using auto in silico consensus inverse docking (ACID) server. Among the seven selected 1,5- benzothiazepine derivatives, consensus inverse docking (CID) against PDB-binding database identified potential therapeutic targets using structure-based screening. The comparative analysis predicted 4-hydroxyphenylpyruvate dioxygenase (HPPD) (Uniprot ID: P32754), involved in the regulation of blood tyrosine levels by catalyzing the reaction of HPPD to homogentisic acid in tyrosine catabolism pathway. Dimethylglycine dehydrogenase (DMGDH) (Uniprot ID: Q9AGP8), a flavin adenine dinucleotide (FAD)‐ and tetrahydrofolate (THF)‐dependent, mitochondrial matrix enzyme that degrades choline and transfers electrons to the respiratory chain, one‐carbon metabolism, and folate receptor beta (Uniprot ID: P14207), which is a glycosylphosphatidylinositol-linked protein capturing ligands from the extracellular matrix and passages them inside the cell through endosomal pathway and recognized as an emerging biomarker for cancer and chronic inflammatory diseases, as potential targets for the selected derivatives. These findings warrant for in vitro cell-based experimental validation of the currently identified actives.
Keywords
Pharmacokinetics
1,5-benzothiazepine
Chalcones
Heterogeneous CuO catalyst

1 Introduction
Organic synthesis shows an essential part in the journey of drug discovery. Especially, in development of privileged heterocyclic moieties is a demanding area of research in medicinal chemistry. S and N-containing heterocycles such as thiazepine and its derivatives are significant structural scaffolds of seven-membered heterocycles that exhibit a broad spectrum of biological activity (Struga et al., 2009). The unique characteristic of thiazepine moiety is that it is vital against various families of targets and creating them an interesting heterocyclic ring system (Tailor et al., 2014).
The therapeutic candidates of benzothiazepines like “thiazesim” is a well-known anti-depressant in the pharmaceutical market followed by diltiazem, siratiazem and clentiazem as a calcium channel blocker along with clothiapine and quetiapine as anti-psychotic drug drugs of this family.
Benzothiazepine is formed when thiazepine is fused with a benzene ring. Benzothiazepines are associated with many pharmacological activities such as anticonvulsant, antifungal, antioxidant, antimicrobial, anti-inflammatory, antiarrythmic, antipsychotic drug activity, anti-HIV activity, anticancer activity, etc (Devi et al., 2020).
Chalcone is an α,β-unsaturated ketone which has diversity of pharmacological activities such as anti-tumor (Abbas et al., 2014), anti-inflammatory (Iqbal et al., 2014), antiulcer (Tanaka et al., 2009) and antibacterial (Parikh and Joshi, 2013). Chalcones have also been reported to show nematicidal (Awasthi et al., 2009), larvicidal (Begum et al., 2011) and insect antifeedant (Devi et al., 2021) activities. Many methods have been reported for the formation of chalcones such as by reacting aromatic aldehydes and phenyl acetylene to mediate Favorskii reaction in the occurrence of Montmorillonite KSF (Ameta et al., 2022), condensation reaction of acetophenones with aromatic aldehydes in occurrence of aqueous alkali (Prasad et al., 2007) and via microwave induced method (Ameta et al., 2011). Chalcones are used as starting substrate for synthesizing different sized bioactive heterocycles. In the present study, chalcones and 2-aminothiophenol were used as main precursors for the synthesis of the title product using CuO nanocatalyst.
Over the past few decades, the use of catalyst in chemical reaction has gained significant attention in both pharmaceutical and specialty chemical industries due to their useful properties such as environment friendly production technology, efficient recovery, excellent activity, low preparation cost, high selectivity, great stability and good recyclability. Transition metal oxides are an important class of semiconductors and display both Lewis acid and base at their surface. CuO offers low coordinating sites and greater surface area that makes it very reactive and help in catalyzing carbon heteroatom and carbon–carbon bond formation (Ranu et al., 2012).
A chemical compound’s pharmacological profiling generates a fundamental selectivity outline that symbolizes its promiscuity towards variety of biological targets (Furlan et al., 2018). Taking the advantage of multi-faced nature of active pharmacological compounds, inverse molecular docking was introduced (Grinter et al., 2011). This technique has been successfully utilized for identifying biological target(s) for synthesized compounds among a family of receptors. Moreover, this strategy enables early expectation of drugs side-effects and toxicity in response to array of biological targets (Rollinger, 2009). Moreover, time complexity of inverse docking has been reduced by focusing on docking search space to compute binding scores (Bissantz et al., 2004).
Keeping the diverse and broad pharmacological action of benzothiazepines in view and as part of our ongoing study on the development of new bioactive heterocycles combining nitrogen and sulphur atoms (Ameta et al., 2011), a sustainable method for converting chalcones into 1,5-benzothiazepines is detailed in this study. Herein we report the preparation of 1,5-benzothiazepine derivatives by condensation of substituted chalcones and 2- aminothiophenol using CuO nanocatalytic framework as a time saving, reusable and nontoxic catalyst. Further, in-silico pharmacokinetic properties have been evaluated for these newly synthesized 1, 5- benzothiazepine derivatives to explore for future drug discovery pipeline. Advanced high-computing inverse docking strategy has been utilized for prioritizing the potential biological targets against 1,5-benzothiazepine derivatives and the derived results have been subjected to validation by using molecular dynamics simulation platforms for precise drug designing modules.
2 Materials and methods
All chemicals were acquired from Merck (USA) and Sigma-Aldrich (USA) of analytical grade and were utilized without additional purification. Melting points of the synthesized compounds were dictated using melting point apparatus in open glass capillaries. To observe the achievement of the reaction, thin layer chromatography (TLC) was accomplished via silica gel pre-coated aluminium sheets (Merck 60–120). 1H NMR spectra were recorded at 500 MHz and 600 MHz in DMSO and CDCl3 respectively while 13C NMR were detailed at 100 MHz in DMSO and CDCl3 on FT NMR spectrometer Bruker Advance NEO (in δ ppm unit) using TMS (tetramethylsilane) as an internal standard. The protons were denoted as 1H: s (singlet), t (triplet) and m (multiplet).
2.1 Synthesis of chalcones (3a-n)
Equimolar quantities 4-hydroxyacetophenone (0.01 mol) and substituted aryl aldehydes (0.01 mol) were put in ethanol (30 mL) and agitated at room temperature. With constant stirring, aqueous solution of KOH (15 mL, 40 %) was put to the reaction combination. The reaction combination was retained overnight and transferred onto ice water and diluted with dilute HCl. The solid acquired was strained off and recrystallized from ethanol to obtain chalcones.
2.2 Synthesis of 1,5- benzothiazepine derivatives (5a-n)
To a minimum quantity of ethanol, substituted chalcones 1 (0.01 mol) were dissolved. To this, 2- aminothiophenol 2 (0.01 mol) and a pinch of CuO catalyst were put in and the resulting reaction combination was refluxed for 3 h at 60 °C. Afterwards, the reaction mixture was acidified by the addition of glacial acetic acid (5–6 drops) and reflux was continued for further 2–3 h. The achievement of the reaction was examined by TLC [Benzene: Ethylacetate; 9:1 v/v]. The reaction combination was permitted to cool at room temperature and the content was transferred against ice water. The mixture was filtered off and solid obtained was purified by column chromatography to afford the title product [(Supplementary Information: Appendix III (5a-n)].
2.3 In silico pharmacokinetic analysis of 1,5- benzothiazepine derivatives
Molinspiration server (https://www.molinspiration.com) was employed for analyzing the drug likeliness and molecular descriptors of synthesized 1,5- benzothiazepine derivatives. As per Lipinski’s rules of five, a drug like molecule must have logP ≤ 5, molecular weight ≤ 500, number of hydrogen bond acceptors ≤ 10, and the number of hydrogen bond donors ≤ 5. And, violation of any of the above rule may affect the oral bioavailability.
where, TPSA = Total Polar Surface Area.
The prediction of the bioactivity score of drugs was performed by calculating vital molecular descriptors such as logP, polar surface area, number of hydrogen bond donors and acceptors. Further, ADMET profiling (Absorption, Distribution, Metabolism, Excretion and the Toxicity) of derivatives was performed using admetSAR (Bhardwaj et al., 2019; Khan et al., 2015). admetSAR is a web-base tool containing manually curated data of various known chemical compounds and serve as a controller for determining ADMET properties of novel input compounds sharing structural and functional similarity.
2.4 Potential therapeutic target identification
Systematic identification of potential therapeutic targets for 1,5- benzothiazepine derivatives was performed by using in silico consensus inverse docking (ACID) server. ACID is a consensus platform is capable of figuring the binding energies of docked complexes from AutoDock Vina, LEDOCK, PLANTS, and PSOVin (Wang et al., 2019). In order to expand the accuracy of the docked pose and binding mode prediction, a system level drug repurposing task was performed by integrating Molecular Mechanics/Poisson–Boltzmann Surface Area (MM/PBSA) and X-SCORE for concluding binding energy calculation (Singh et al., 2017). This includes following steps:
-
Selection of test dataset containing 16,151 protein–ligand entries from PDB-bind database;
-
Preparation of 3D structure of proteins by removal of water molecules using PDB2PQR 1.6 21 program;
-
Automated active site prediction of target proteins followed by the generation of hundred conformations for every ligand versus target protein’s active site;
-
Calculation of binding free energy (ΔGbind) via MM/PBSA and X-score methods, under below equation:
2.5 Molecular dynamics (MD) simulation
MD simulation of the stated docked complexes was supported by the usage of GROMACS package. In order to research the conformational balance, MD simulations have been done by following the same old tactics. For the era of ligand topologies and random human error elimination, the ACPYPE device was used. By employing the single factor price (SPC) water model and the inclusion of water molecules, a solution was created. Ordered water molecules serve as a guide for protein–ligand binding affinity by bridging the interaction between the two (Khan et al., 2018; Khan et al., 2018). Complete neutralization was carried out by adding of counter ions (Na+ and Cl-) at 0.15 M of biological salt awareness. A particle-mesh-based Ewald (PME) method was used to calculate the electrostatic force and strength, which helps to assign particular periodic boundary conditions (Gupta et al., 2020). Lennard-Jones interactions cut-off was set to 10 to limit the motion of all covalent bonds pertaining to hydrogen atoms. To investigate the MD simulations' paths toward electricity minimization, a six-step approach was used (a) using the Broyden-Fletcher-Goldfarb-Shanno (LBFGS) set of principles, solvent molecule energy minimization prior to the device as a whole, (b) To reach the equilibrium state, non-hydrogen solute atoms were limited to 300 K temperature and 1 bar for 40 ns, (c) Initial simulations were conducted at controlled temperatures and pressures using Berendsen thermostats and a set of barostat rules, (d) The study of the root-mean-square (RMSD) deviation is aided by the initial trajectory information, (e) root-mean-square fluctuations (RMSF), and (f) radius of gyration (Rg) for protein–ligand complexes (Anwer et al., 2020).
3 Results
3.1 Synthesis of CuO catalyst, chalcones and 1,5-benzothiazepines
Our research aim was to develop an efficient methodology for the production of 1,5-benzothiazepine derivatives, which is a seven membered heterocyclic compound that contains N- and S-atoms.
Firstly, the desired heterocyclic CuO catalyst was prepared by using a reported method (Luna et al., 2015). The synthesized catalyst was categorized by SEM and XRD techniques. The SEM images and XRD pattern are presented in Figure S-1 (Supplementary Information).
Through Claisen-Schmidt condensation reaction, the chalcones were prepared by reacting p-hydroxyacetophenone with various substituted aldehydes in a basic medium (KOH). The goal compounds (5a-n) were produced by Michael addition of 2-aminothiophenol to chalcones in acidic medium in the presence of CuO catalyst (Scheme S-1 and Appendix- I, II and III; Supplementary Information).
3.2 In silico pharmacokinetic analysis of 1,5- benzothiazepine derivatives
Pharmacokinetic parameters such as CLogP, lipophilicity, molecular weight and topological polar surface area (TPSA) of the newly synthesized 1, 5-benzothiazepine derivatives were computed using Molinspiration server and are summarized in Table 1. Abbreviations: MiLogP: octanol/water partition coefficient; TPSA: topological polar surface area; natoms: number of atoms; MW: molecular weight; nON: number of hydrogen bond acceptors; nOHNH: number of hydrogen bond donors; nviolations: number of Lipinski’s rule-of-five violations; nrotb: number of rotatable bonds. According to Lipinski’s rule-of-five, for a compound to be drug-like, it should have ≤5 hydrogen bond donors, ≤10 hydrogen bond acceptors, molecular weight ≤500 and Milogp ≤ 5. Violation of two or more rule may result in reduced bioavailability.
Compounds
miLogP
TPSA
natoms
MW
nON
nOHNH
nviolations
nrotb
volume
5a
-0.24
181.61
32
377.46
10
7
1
2
377.44
5b
0.57
153.01
26
349.42
8
2
0
7
317.46
5c
1.82
156.71
23
347.43
6
2
0
4
287.55
5d
1.22
179.56
28
361.46
11
2
1
5
335.46
5e
0.39
179.73
43
345.46
10
1
1
6
339.08
5f
-1.34
156.76
28
391.48
9
7
1
4
411.27
5g
-0.68
172.47
34
365.88
13
4
2
10
421.91
5h
1.78
169.04
43
410.33
9
4
2
11
442.87
5i
1.13
188.62
32
400.32
12
1
0
5
225.56
5j
0.87
198.45
31
332.42
11
2
1
6
267.87
5k
-0.43
181.61
32
365.88
10
7
1
2
377.79
5l
-1.32
165.43
30
365.88
11
2
1
4
569.23
5m
-1.26
172.07
27
331.43
8
5
1
4
387.43
5n
1.97
198.55
24
347.43
7
2
4
5
367.43
3.3 Molecular dynamic (MD) simulation
Among seven selected 1,5- benzothiazepine derivatives (5b, 5d, 5f, 5 h, 5j, 5 k, and 5 l), consensus inverse docking against PDB-binding database identified potential therapeutic targets using structure-based screening through ACID server (Table 3).
Compounds
Targets
Binding Energy (kcal/mol)
PDB Code
Uniprot ID
Name
5a
CASP3
TARS
FOL1-10.77
-9.21
-9.012XYG
4HWT
2BMBP42574
P26639
P53848Caspase-3
Threonine--tRNA ligase, cytoplasmic
Folic acid synthesis protein FOL1
5b
HPPD
DMGDH
HNMT-10.01
-9.22
-8.763ISQ
1PJ5
2AOTP32754
Q9AGP8
P501354-hydroxyphenylpyruvate dioxygenase
Dimethylglycine dehydrogenase
Histamine N-methyltransferase
5c
TPMT
DMGDH
GRIK2-11.65
-10.54
-10.112BZG
1PJ5
5CMMP51580
Q9AGP8
Q13002Thiopurine S-methyltransferase
Dimethylglycine dehydrogenase
Glutamate receptor ionotropic, kainate 2
5d
HPPD
PGR
DMGDH-9.83
-9.21
-8.453ISQ
1SQN
1PJ5P32754
P06401
Q9AGP84-hydroxyphenylpyruvate dioxygenase
Progesterone receptor
Dimethylglycine dehydrogenase
5e
HPPD
PGR
THRA-9.76
-8.22
-8.063ISQ
1SQN
3ILZP32754
P06401
P108274-hydroxyphenylpyruvate dioxygenase
Progesterone receptor
Thyroid hormone receptor alpha
5f
HPPD
FOLR2
DMGDH-10.76
-10.05
-9.233ISQ
4KN0
1PJ5P32754
P14207
Q9AGP84-hydroxyphenylpyruvate dioxygenase
Folate receptor beta
Dimethylglycine dehydrogenase
5g
HPPD
LTA4H
MT-CO2-9.65
-9.01
-8.793ISQ
3U9W
3VRJP32754
P09960
P004034-hydroxyphenylpyruvate dioxygenase
Leukotriene A-4 hydrolase
Cytochrome c oxidase subunit 2
5h
HPPD
MT-CO2
DMGDH-10.65
-10.04
-9.563ISQ
3VRJ
1PJ5P32754
P00403
Q9AGP84-hydroxyphenylpyruvate dioxygenase
Cytochrome c oxidase subunit 2
Dimethylglycine dehydrogenase
5i
HPPD
P2RY12
FOLR2-10.58
-10.07
-9.663ISQ
4PXZ
4KN0P32754
Q9H244
P142074-hydroxyphenylpyruvate dioxygenase
P2Y purinoceptor 12
Folate receptor beta
5j
HPPD
LTA4H
FOLR2-11.76
-10.32
-10.213ISQ
3U9W
4KN0P32754
P09960
P142074-hydroxyphenylpyruvate dioxygenase
Leukotriene A-4 hydrolase
Folate receptor beta
5k
HPPD
CHRM4
FOLR2-11.56
-10.34
-10.033ISQ
5DSG
4KN0P32754
P08173
P142074-hydroxyphenylpyruvate dioxygenase
Muscarinic acetylcholine receptor M4
Folate receptor beta
5l
HPPD
LTA4H
FOLR2-9.87
-9.62
-9.053ISQ
3U9W
4KN0P32754
P09960
P142074-hydroxyphenylpyruvate dioxygenase
Leukotriene A-4 hydrolase
Folate receptor beta
5m
HPPD
DMGDH
THRA-11.76
-10.32
-10.083ISQ
1PJ5
3ILZP32754
Q9AGP8
P108274-hydroxyphenylpyruvate dioxygenase
Dimethylglycine dehydrogenase
Thyroid hormone receptor alpha
5n
HPPD
DMGDH
TPMT-9.45
-9.01
-8.123ISQ
1PJ5
2BZGP32754
Q9AGP8
P515804-hydroxyphenylpyruvate dioxygenase
Dimethylglycine dehydrogenase
Thiopurine S-methyltransferase
4 Discussion
In this reported method, CuO catalyst which has high surface area shows a vital role to fasten the rate of reaction. The enhanced catalytic activity is due to the catalyst's surface area, which considerably increases as its size decreases. Through Lewis acid site (Cu2+) associated with the enone functional group, a CuO catalyst speeds the Michael addition type coupling. CuO catalysts can also deprotonate the N—H bond in the presence of a Lewis base (O2–) by activating 2-aminothiophenol at the same time. Thus, 1,5- benzothiazepine derivatives were formed by the activation of the reactants through both Lewis acid and basic sites of CuO catalyst as a result of nucleophilic substitution by heterocyclic amine. 1H NMR spectrum of chalcones indicated the existence of doublet at δ 7.03–7.44 ppm and δ 7.35–7.80 ppm corresponding to α-hydrogen and β-hydrogen respectively (Supplementary Information: spectrum of 3d is attached in appendix I). Michael addition reaction of these chalcones to 2-aminothiophenol resulted in the formation of 1,5-benzothiazepines which is confirmed by the spectral studies. Thus in 1H NMR spectrum of 1,5-benzothiazepines, a doublet at δ 3.24–4.08 ppm and a triplet at δ 4.10–4.33 ppm corresponding to –CH2 and –CH groups respectively is observed due to the cyclization of enone system of chalcones (Supplementary Information: spectra of 5e and 5f is attached in appendix II).
Further the reaction conditions were checked by several reaction factors such as catalyst quantity, temperature and solvent. The quantity of the catalyst was determined by varying mol% of CuO catalyst from 5 to 30 mol% (Table S-1; Supplementary Information).
It was found that 20 % is enough for the reaction; further increase in catalyst quantity didn’t increase the yield and reaction time. The reaction didn’t not take place at room temperature, thus chalcones stay unconsumed. The reaction was carried out at different temperature from 40 to 80 °C (Table S-2; Supplementary Information) and it was noticed that the yield of the product was maximum at 60 °C, thus it was considered as the optimum reaction temperature.
In the lack of solvent, the reaction gave very low yield (Table S-3; Supplementary Information). Among different solvents considered, ethanol was observed to be the finest solvent providing highest yield of the product.
Recyclability and reusability of the catalyst are important aspect in organic synthesis. Hence, the product is extracted using a solvent from the reaction mixture. The catalyst CuO was recovered by using ethyl acetate. The aqueous layer having CuO particles was isolated by simple purification and washed with ethyl acetate and used for the next three successive runs without any extensive loss of the catalytic activity (Figure S-2; Supplementary Information).
4.1 Analysis of in silico pharmacokinetic properties
The sum of fragment-based contribution and correlation factors are termed as CLogP = octanol/water partition coefficient. Based on Lipinski’s rule of five, compounds having molecular weight ranging between (292–470 i.e., > 500) were selected as low molecular weight compounds and can be easily diffused, transported and absorbed in comparison with heavy compounds. If a chemical becomes too bulky, it can interfere with the therapeutic effects of drugs. By screening molecules with hydrogen bond acceptors and donors fewer than 10 and 5, respectively, it was possible to prioritise the compounds. The permeability of compounds was assessed by computing logP value less than 5. This allows easy penetration of drugs through biomembranes. The computation of topological polar surface area (TPSA) based on O- and N- centered polar fragments assists in characterization of drug and intestinal absorption, bioavailability, Caco-2 penetrability and blood–brain barrier diffusion. Both, the degree of lipophilicity and TPSA indicate strong interaction of drug with membranes and hydrogen bonding potential of the compound. Higher quantity of rotatable bonds raises the flexibility of molecule and resultantly adaptability for effective interactions within binding pocket. Whereas, admetSAR enables the computation of absorption, distribution, metabolism, excretion and toxicity (ADMET) properties. For entire derivatives, blood–brain barrier (BBB) permeation, HIA (human intestinal absorption), Caco-2 cell permeability and AMES test (a type of biological assay for assessing the mutagenic potential of chemical compounds) were considered and shortened in Table 2.
Compounds
Absorption
Metabolism
Excretion
Toxicity
P-Glycoprotein
CYP-450 Substrate
CYP-450 Inhibitor
BBB
HIA
Pg-S
Pg-I
2C9
2D6
3A4
1A2
2C9
2D6
2C19
3A4
ROCT
AMES
Carcinogen
5a
+
+
-
-
-
-
-
-
-
-
-
-
-
+
-
5b
+
+
-
-
-
-
-
-
-
-
-
-
-
+
-
5c
+
+
-
-
-
-
-
+
-
-
-
-
-
-
-
5d
+
+
-
-
-
-
-
-
-
-
-
-
-
-
-
5e
+
+
-
-
-
-
-
+
-
-
-
-
-
-
-
5f
+
+
-
-
-
-
-
-
-
-
-
-
-
-
-
5g
+
+
-
-
-
-
-
-
-
-
-
-
-
-
-
5h
+
+
-
-
-
-
-
-
-
-
-
-
-
-
-
5i
+
+
-
-
-
-
-
-
-
-
-
-
-
-
-
5j
+
+
-
-
-
-
-
-
-
-
-
-
-
-
-
5k
+
+
-
-
-
-
-
-
-
-
-
-
-
-
-
5l
+
-
-
-
-
-
-
-
-
-
-
-
-
-
-
5m
+
+
-
-
-
-
-
+
-
-
-
-
-
-
-
5n
+
+
+
-
-
-
-
-
+
-
-
-
-
-
-
For efficient drug absorption and allowance in the liver, concentration of several isoforms of cytochrome P450 super family (i.e., CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4) were estimated. This identifies that ROS acts as a substrate for P-glycoprotein that supports the efflux of drug before metabolism leads to therapeutic drug failure and metabolic clearance. Based on predicted values of admetSAR, seven ligands derived from 1,5- benzothiazepine (5b, 5d, 5f, 5 h, 5j, 5 k, and 5 l) were able to penetrate blood–brain barrier permeation, Caco-2 cell and human intestine and showed no toxicity or mutagenic effect.
4.2 Inverse molecular docking
Comparative analysis revealed 4-hydroxyphenylpyruvate dioxygenase (Uniprot ID: P32754), dimethylglycine dehydrogenase (Uniprot ID: Q9AGP8) and folate receptor beta (Uniprot ID: P14207) as major targets for the selected 1,5- benzothiazepine derivatives. In addition to the above stated targets, muscarinic acetylcholine receptor M4; leukotriene A-4 hydrolase; cytochrome c oxidase subunit 2; progesterone receptor; histamine N-methyltransferase were also screened as potential targets. In case of 4-hydroxyphenylpyruvate dioxygenase (HPPD), 5j and 5 k served as potential inhibitors with binding energies of −11.76 kcal/mol and −11.56 kcal/mol respectively. For dimethylglycine dehydrogenase (DMGDH), 5b and 5d showed inhibiting activity with binding energies of −9.22 kcal/mol and −10.54 kcal/mol respectively. For folate receptor beta, 5f and 5j computed binding energies of −10.05 kcal/mol and −10.32 kcal/mol respectively.
4.3 Description of main targets for the selected 1,5- benzothiazepine derivatives
4-hydroxyphenylpyruvate dioxygenase (HPPD): It is a Fe (II)-containing oxygenase enzyme involved in catabolic transformation of 4-hydroxyphenylpyruvate into homogentisate in tyrosine metabolism. Literature citation revealed linkage of HPPD with metabolic disorders such as alkaptonuria and Type III tyrosinemia (Yang, 2003). Being involved in industrial production of necessary co-factors in plants related to herbicide development, HPPD inhibitors have equal importance in agribiotechnology. High level of tyrosine concentration in blood due to lower HPPD enzyme concentration in human intestine results in lenient mental retardation at birth and degradation in vision (Hühn et al., 1998). Therefore, HPPD is considered as a potential goal for drug and pesticide finding (Lin et al., 2019). Docking analysis revealed that Phe424, Asn423, Tyr456, Glu394, Asn282, Gln307 and Ser267 play a significant part in the binding of C-3 position of hydroxyl group in the benzene ring and sulfur group of both substrate residues (Figure S-3 a,b; Supplementary Information).
Dimethylglycine dehydrogenase (DMGDH): It is a mitochondrial matrix flavoprotein, which is active in choline and 1-carbon metabolism. It is involved in catalytic demethylation of dimethylglycine to form sarcosine that bounds to FAD cofactor in oxidative phosphorylation machinery. Higher level of DMGDH in blood stream is accountable for metabolic disorders related to choline metabolism (Binzak et al., 2001). Docking studies revealed that amino acid residues in active ATP binding sites (GLU 915, ARG 927, LEU 868, GLY 920, GLU 883, LYS 885, PHE 916, PHE 919, LYS 1053 and ASN 921) are hydrogen bonded to methoxy group at C3 and C9 position of 1,5- benzothiazepine derivatives. The presence of van der Waals interactions with Met 68, Val 69 and Leu 90 with C4 group of benzene ring are important stuctural requirenent for anti-toxicity, which was also found during the analysis (Figure S-3c,d; Supplementary Information).
Folate receptor beta (FR): It is a glycosylphosphatidyinositol-linked protein capturing ligand from the extracellular matrix and carries them inside the cell through endosomal pathway (Yi, 2016). It binds to folate and reduces folic acid derivatives mediating 5-methyltetrahydrofolate inside the cell. It is considered as a potential tumor biomarker. High expression level of folate receptor (FR) occurs in human malignancies, ovarian, endometrial and colon cancer. The FR is an emerging therapeutic target for the cure of chronic inflammatory diseases and cancer (Chandrupatla et al., 2019). The results of docking showed that the major H-bonding interaction with benzene ring (C-9th position of –OH group and the phenyl ring D) could associate with the (O-atom) key chain of Tyr88, Phe86, Asn124, Gln165 (Figure S-3 e,f; Supplementary Information). Tyr88 and Phe86 were found to be the main active residues accountable for hindrance of folate receptor beta.
The firmness of protein–ligand complexes was further examined by molecular dynamics simulations. The backbone of initial structure of the protein target was utilized for the generation of root mean square deviation (RMSD) graphs with respected to time at 40 ns with an average RMSD computed as 0.334 ± 0.05 nm (Ahmed et al., 2020). In case of 4-hydroxyphenylpyruvate dioxygenase Fig. 1 (a,b,c) and dimethylglycine dehydrogenase Fig. 2 (a,b,c) related complexes, RMSD values gained till 10 ns and conjunction was detected from 40 ns but slight variations stayed throughout. The overlaid graphical demonstration of time-dependent RMSD of protein–ligand complexes are represented in Fig. 1(a), Fig. 2(a) and 3(a). The dictionary of secondary structure of proteins (DSSP) suggests that active sites lies in the coil region and the majority of binding sites shaped β-helix structure. Residue-based root mean square fluctuations (RMSF) study of protein–ligand docked complexes was accomplished to know the flexibility of each residue and represented in Fig. 1(c), Fig. 2(c) and 3(c). RMSF values for all the docked complexes had large fluctuations (0.33–1.67 nm) for initial 20 residues in each case due to unavailability of structural information of the target protein. Further, lesser fluctuations at binding and active site indicate rigidity and intactness of the binding cavity. gmx_gyrate calculates Rg values indicating solidity and structural alterations of the docked complexes. Additionally, it calculates the atomic mass that corresponds to the complexe’s centres of mass. Please refer, Fig. 1(b), Fig. 2(b) and 3(b). Average Rg values of 4-hydroxyphenylpyruvate dioxygenase, dimethylglycine dehydrogenase and folate receptor beta ranged between (2.36–2.66 nm), (2.29–2.89 nm) and (2.44–2.56 nm) respectively with no fluctuations after 25000 ps. Further, Rg values correlate with RMSD values of backbone Cα atoms validating the firmness of the prioritized protein ligand complexes. This justifies the suitability of the prioritized 1,5- benzothiazepine derivatives to be further explored as potential drugs against various human disorders.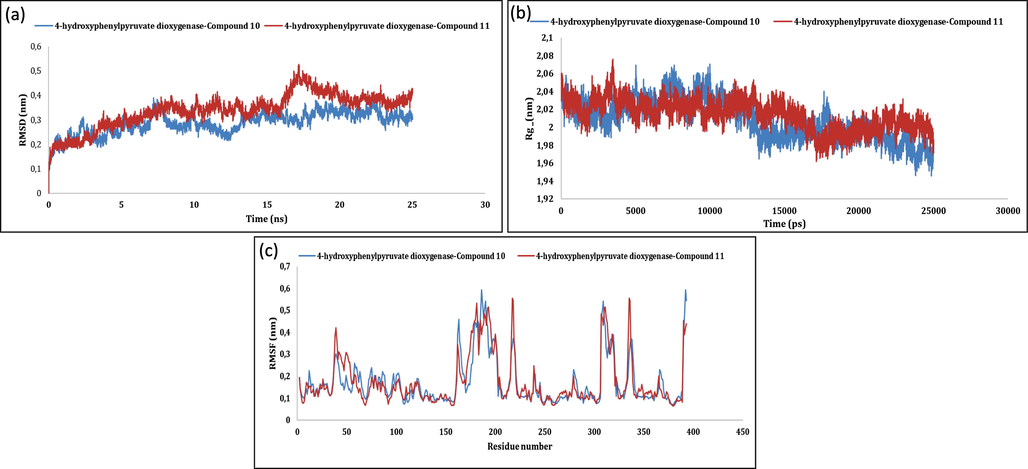
MD simulation of 4-hydroxyphenylpyruvate dioxygenase-5j and 4-hydroxyphenylpyruvate dioxygenase-5 k: (a) RMSD; (b) Rg; (c) RMSF.
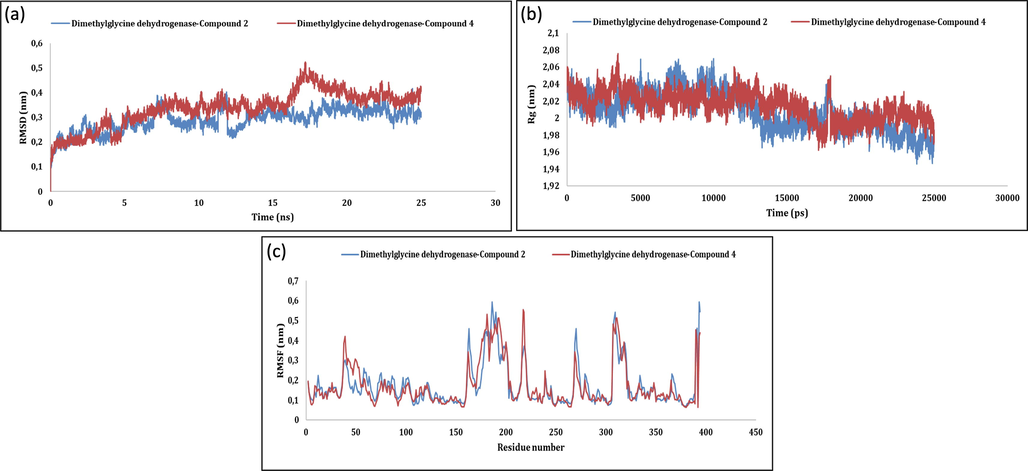
MD simulation of Dimethylglycine dehydrogenase-5b and dimethylglycine dehydrogenase-5d: (a) RMSD; (b) Rg; (c) RMSF.
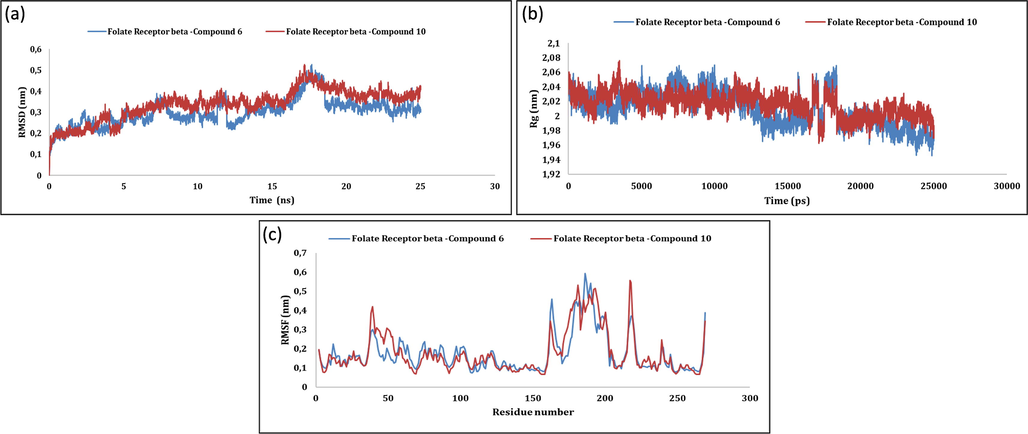
MD simulation of folate receptor beta −5f and folate receptor beta −5j: (a) RMSD; (b) Rg; (c) RMSF.
5 Conclusion
An improved synthetic route was created for the synthesis of 1,5-benzothiazepines using CuO nanocatalyst, which is recyclable, non-hazardous, inexpensive, commercially available catalyst and works under mild reaction conditions. Further, in silico pharmocokinetics of the selected (newly synthesized) 1,5- benzothiazepines based on consensus inverse docking against PDB-Binding database identified potential therapeutic targets using structure-based screening through auto consensus inverse docking (ACID) server was studied. Comparative analysis predicted 4-hydroxyphenylpyruvate dioxygenase (Uniprot ID: P32754), dimethylglycine dehydrogenase (Uniprot ID: Q9AGP8) and folate receptor beta (Uniprot ID: P14207) as major targets for the selected 1,5- benzothiazepine derivatives. The current findings demand endorsement via in vitro cell-based experimental studies using these newly synthesized 1,5- benzothiazepine derivatives to be further explored as potential drug molecules.
Acknowledgments
The authors are thankful to the Department of Chemistry, Sardar Patel University, Vallabh Vidyanagar for providing necessary facilities. The authors also extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through the Project No. RUP-5.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Synthesis and cytotoxicity studies of 4-alkoxychalcones as new antitumor agents. J. Mater. Environ. Sci.. 2014;51:281-292.
- [Google Scholar]
- Identifying novel inhibitor of quorum sensing transcriptional regulator (SdiA) of Klebsiella pneumoniae through modelling, docking and molecular dynamics simulation. J. Biomol. Struct. Dyn. 2020
- [CrossRef] [Google Scholar]
- Synthesis and in vitro anti breast cancer activity of some novel 1, 5-benzothiazepine derivatives. J. Serb. Chem. Soc.. 2011;77:725-731.
- [CrossRef] [Google Scholar]
- Microwave induced improved synthesis of some novel substituted 1, 3-diarylpropenones and their antimicrobial activity. EJ-CHEM.. 2011;8:665-670.
- [Google Scholar]
- Montmorillonite KSF mediated Favorskii reaction based eco-friendly fabrication of some 1,3-diarylpropenones using phenyl acetylene and aromatic aldehydes. J. Iran. Chem. Soc.. 2022;19:3943-3949.
- [CrossRef] [Google Scholar]
- Unravelling the interaction of glipizide with human serum albumin using various spectroscopic techniques and molecular dynamics studies. J. Biomol. Struct. Dyn. 2020
- [CrossRef] [Google Scholar]
- Antifilarial activity of 1,3-diarylpropen-1-one: effect on glutathione-Stransferase, a phase II detoxification enzyme. Am. J Trop. Med. Hyg.. 2009;80:764-768.
- [Google Scholar]
- Mosquito larvicidal studies of some chalcone analogues and their derived products: structure–activity relationship analysis. Med Chem. Res.. 2011;20:184-191.
- [Google Scholar]
- Comparative assessment of the therapeutic drug targets of C. botulinum ATCC 3502 and C. difficile str. 630 using in silico subtractive proteomics approach. J. Cell. Biochem.. 2019;120:16160-16184.
- [Google Scholar]
- Cloning of dimethylglycine dehydrogenase and a new human inborn error of metabolism, dimethylglycine dehydrogenase deficiency. Am. J. Hum. Genet.. 2001;68:839-847.
- [Google Scholar]
- High-throughput modeling of human G-Protein coupled receptors: amino acid sequence alignment, three-dimensional model building, and receptor library screening. J. Chem. Inf. Comput. Sci.. 2004;44:1162-1176.
- [Google Scholar]
- The folate receptor β as a macrophage-mediated imaging and therapeutic target in rheumatoid arthritis. Drug Deliv. Transl. Res.. 2019;9:366-378.
- [Google Scholar]
- Bis- and mono-substituted chalcones exert anti-feedant and toxic effects on fall armyworm Spodoptera frugiperda. Saudi J. of Biol. Sci. 2021
- [CrossRef] [Google Scholar]
- Recent advances in the synthetic chemistry of 1,5 -benzothiazepines : A minireview. J. Heterocycl. Chem. 2020
- [CrossRef] [Google Scholar]
- Inverse molecular docking as a novel approach to study anticarcinogenic and anti-neuroinflammatory effects of Curcumin. Molecules. 2018;23:3351.
- [CrossRef] [Google Scholar]
- An inverse docking approach for identifying new potential anti-cancer targets. J. Mol. Graph. Model.. 2011;29:795-799.
- [CrossRef] [Google Scholar]
- Structure-based screening of non-β-lactam inhibitors against class D β-lactamases: an approach of docking and molecular dynamics. ACS Omega. 2020;516:9356-9365.
- [Google Scholar]
- Novel and recurrent tyrosine aminotransferase gene mutations in tyrosinemia type II. Hum. Genet.. 1998;102:305-313.
- [Google Scholar]
- Synthesis, anti-inflammatory and antioxidant activity of ring-a-monosubstituted chalcone derivatives. Med. Chem. Res.. 2014;23:4383-4394.
- [Google Scholar]
- Implication of caspase-3 as a common therapeutic target for multineurodegenerative disorders and its inhibition using nonpeptidyl natural compounds. BioMed. Res. Int.. 2015;379817:1-9.
- [Google Scholar]
- Inhibition of C298S mutant of human aldosereductase for antidiabetic applications: Evidence from in silico elementary mode analysis of biological network model. J. Cell. Biochem.. 2018;119:6961-6973.
- [Google Scholar]
- Aspartate-β-semialdehyde dehydrogenase as a potential therapeutic target of Mycobacterium tuberculosis H37Rv: Evidence from in-silico elementary mode analysis of biological network model. J. Cell. Biochem.. 2018;119:2832-2842.
- [Google Scholar]
- Molecular insights into the mechanism of 4-hydroxyphenylpyruvate dioxygenase inhibition: enzyme kinetics, X-ray crystallography and computational simulations. FEBS J.. 2019;286:975-990.
- [Google Scholar]
- Preparation and characterization of copper oxide nanoparticles synthesized via chemical precipitation method. OALib Journal. 2015;2:1-8.
- [CrossRef] [Google Scholar]
- Antibacterial and antifungal screening of newly synthesized benzimidazole-clubbed chalcone derivatives. Med. Chem. Res.. 2013;22:3688-3697.
- [Google Scholar]
- Synthesis and biological evaluation of some novel chalcone derivatives. Oriental J. Chem.. 2007;23:927-937.
- [Google Scholar]
- Copper nanoparticle-catalyzed carbon-carbon and carbon-heteroatom bond formation with a greener perspective. Chemsuschem.. 2012;5:22-44.
- [Google Scholar]
- Accessing target information by virtual parallel screening— The impact on natural product research. Phytochem. Lett.. 2009;2:53-58.
- [Google Scholar]
- Mechanistic principles behind molecular mechanism of rifampicin resistance in mutant RNA polymerase beta subunit of Mycobacterium tuberculosis. J. Cell. Biochem.. 2017;118:4594-4606.
- [Google Scholar]
- Synthesis, pharmacological and antiviral activity of 1,3-thiazepine derivatives. Eur. J. Med. Chem.. 2009;44:4960-4969.
- [Google Scholar]
- Synthesis, characterization and antimicrobial activity of 2-(11-oxodibenzo [b, f][1,4]thiazepin-10(11H)-yl)-N (substituted phenyl) acetamide derivatives. Indian J. of Chem. -Section B. 2014;53:1263-1268.
- [Google Scholar]
- Sofalcone, an anti-ulcer chalcone derivative, suppresses inflammatory crosstalk between macrophages and adipocytes and adipocyte differentiation: implication of heme-oxygenase-1 induction. Biochem. Biophys. Res. Commun.. 2009;23:566-571.
- [Google Scholar]
- ACID: a free tool for drug repurposing using consensus inverse docking strategy. J. Cheminformatics. 2019;11:73.
- [CrossRef] [Google Scholar]
- 4-Hydroxyphenylpyruvate dioxygenase as a drug discovery target. Drug News Perspect.. 2003;16:493-496.
- [CrossRef] [Google Scholar]
- Folate receptor-targeted diagnostics and therapeutics for inflammatory diseases. Immune Netw.. 2016;16:337-343.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2022.102528.
Appendix A
Supplementary material
The following are the Supplementary data to this article:




