

Translate this page into:
Mutational signatures on ischemic stroke-associated genes in Saudi human genome
⁎Corresponding author. alaguraj.veluchamy@kaust.edu.sa (Alaguraj Veluchamy) alaguraj.v@gmail.com (Alaguraj Veluchamy)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Stroke is a neurological syndrome, and it leads to 5.8 million mortalities worldwide annually. In the Kingdom of Saudi Arabia (KSA), stroke was predicted to have 57%-67% incidence rate against a population growth rate of 12.8%. Current state of the art in stroke research in KSA is limited to epidemiological, prevalence data and there is a lack of genetic basis of stroke among Saudi individuals and their risk for disease traits. Despite the better health care services in KSA, a genetic approach is needed for stroke, as it is a manifestation of both monogenic Mendelian and polygenic disorder. Here, we propose to analyze and annotate Saudi specific genome variations associated with stroke. In this study, we explored the non-coding and genic regions using 28 whole genomes of individuals from Saudi population. We explored stroke susceptible genes for additional variation. Analysis of 49 genes which are stroke-associated for single nucleotide polymorphism (SNPs), obtained from whole genomes, reveals variations in atleast 13 candidate genes. In conclusion, whole genome sequencing and annotation of SNPs in the population of Saudi Arabia provide an insight into genetics of stroke. This analysis furnish a list of probable novel Saudi specific mutations that could be associated with stroke, once a cohort of disease data can beobtained. In addition, we conjecture that, by identifying these mutational signatures, stroke subtype and susceptibility to stroke can be uncovered in the future.
Keywords
Functional annotation
Gene ontology
Genome variation
Ischemic stroke
Single nucleotide polymorphism

1 Introduction
Stroke is a heterogeneous neurological impairment and is the third leading cause of mortality (WHO 2017 Global disease burden data: 12.8 million deaths) and disability worldwide (GBD 2017 Mortality Collaborators, 2018). Although multiple factors including age, sex differences, race, diet, hypertension, smoking, sedentary lifestyle etc. makes one susceptible to stroke, genetics is the most prominent one (Boehme et al., 2017). This neurological syndrome is not a single disease and has multiple types such as Ischemic Stroke (Clots), Hemorrhagic stroke (Bleeds), Transient Ischemic Attack (TIA), Cryptogenic stroke, Brain stem stroke. While Ischemic stroke is more prevalent, Hemorrhagic stroke is highly fatal. Several genetic variation have been associated to with the risks of stroke types or subtypes of Ischemic and Hemorrhagic stroke (Lindgren, 2014). Several challenges in studies and factors that influence the incidence in the Kingdom of Saudi Arabia (KSA) have been reviewed recently (Robert and Zamzami, 2014). One study estimated the stroke incidence rate KSA is 29.8/100,000/year (Al Rajeh et al., 1998). Another study as early as 1993 estimated an annual incidence rate 43.8 per 100,000 (Al Rajeh et al., 1993). Diabetes, Hypertension are some of the major risk factors for Saudi population (El Sayed et al., 1999). Ischaemic stroke was the commonest subtype in all Arab countries which corresponds to 85–90% of all strokes worldwide (Benamer and Grosset, 2009).
Emergence of next generation sequencing methods coupled with technological advances in exome-sequencing and whole genome sequencing techniques serves as a clinical molecular diagnostic testing and prediction of risk of diseases. Genetic basis of disease are explained by the use of genome sequence and mutations such as single nucleotide polymorphisms (SNP) to identify individuals and their risk for disease traits. Genome-wide association studies and several decades of work on stroke genetics on European population created a vast amount of both DNA and RNA sequence data that could be tapped into Saudi nationals.
Mendelian and mitochondrial disorders associated with stroke are well documented (Silliman, 2002). Mutation in single gene HDAC9 could lead to a novel stroke type and this variant increases risk of Ischemic stroke by 42% (Bellenguez et al., 2012). For example, genetic variation in the flanking region of PITX2, ZFHX3 and chromosome 9 short arm region are used for classifying Ischaemic stroke. Mitochondrial genome and stroke are highly interrelated since many mitochondrial disorders are shown to cause stroke related disorders (Anderson et al., 2011). Our OMIM search for stroke shows Ischemic stroke susceptibility loci on genes such as NOS3, F5, AALOX5AP, PRKCH, NOTCH3. Stroke is both single gene and polygenic disorder and availability of comprehensive gene panels for Saudi population can not only provide new avenues for prevention but also improve potential molecular approaches, which may be suitable for the identification of stroke susceptibility genes (Saudi Mendeliome Group, 2015). Similarly frequency of mitochondrial sub haplo groups K affects the risk of IS, hence the Saudi and Arabian peninsula mitochondrial genome should be explored (Abu-Amero et al., 2008).
Despite the better health care services in KSA, a genetic approach is needed for tackling high incidence of stroke as it is both monogenic Mendelian and polygenic disorder. One of the main goals of SHGP (Saudi Human Genome project) is disease prevention. Exploration of non-coding and genic region of the Saudi human genome provides us with novel Saudi specific mutations that needs to be associated with stroke. Also, we conjecture that, by identifying and classifying these mutational signatures, a pan Arabian genetic diversity and susceptibility to stroke can be uncovered. To investigate this, a interdisciplinary approach combined with high-throughput data containing human genomes from middle eastern population to explore stroke associated variations was performed. Whole genome sequencing of humans is one of the unique strategies for identifying variants associated with disease and prediction of risk of certain genetic disorders. The objective of the current study is to provide a comprehensive description of variants in Saudi human genome, in particular mutational signatures on genes associated with ischemic stroke. Upon sequencing a cohort of prospective stroke individuals of Saudi population and comparison to the currently available variant annotation would reveal more on genetics of stroke specific to this population.
2 Methods
2.1 Analysis of whole genome of Saudi individuals using the publicly available data
We have retrieved and utilized the whole genome of individuals of Middle-Eastern population available online (Almarri et al., 2021). From the data containing 137 Middle Eastern human genomes, we have used genome of individuals of Saudi population. Almarri et al. grouped the individuals as group A and group B. We have used a representative individual from both group and pooled the variation together for further analysis. Sequencing samples obtained from SRA were tabulated (Table1). A total of 29 samples are available from Saudi population. Mapping and other analyses were performed against human genome version GRch38.
SI
SRA subject Id
Sample number
Sample group
1
APPG7836393
SAUDI12
SaudiA
2
APPG7836394
SAUDI13
SaudiB
3
APPG7836395
SAUDI14
SaudiB
4
APPG7836403
SAUDI22
SaudiA
5
APPG7836397
SAUDI16
SaudiB
6
APPG7836398
SAUDI17
SaudiA
7
APPG7836399
SAUDI18
SaudiB
8
APPG7836401
SAUDI20
SaudiB
9
APPG7836402
SAUDI21
SaudiB
10
APPG7836396
SAUDI15
SaudiB
11
APPG7836404
SAUDI23
Saudi.o
12
APPG7836405
SAUDI24
SaudiB
13
APPG7836406
SAUDI25
SaudiB
14
APPG7836407
SAUDI26
SaudiB
15
APPG7836408
SAUDI28
SaudiA
16
APPG7850840
SAUDI27
SaudiB
17
APPG7850841
SAUDI29
SaudiB
18
APPG7555949
SAUDI1
SaudiA
19
APPG7555950
SAUDI2
SaudiA
20
APPG7555951
SAUDI3
SaudiA
21
APPG7555952
SAUDI4
SaudiA
22
APPG7555953
SAUDI5
SaudiA
23
APPG7555954
SAUDI6
SaudiB
24
APPG7555955
SAUDI7
SaudiB
25
APPG7555956
SAUDI8
SaudiB
26
APPG7555957
SAUDI9
SaudiB
27
APPG7623350
SAUDI10
SaudiA
28
APPG7623351
SAUDI11
SaudiA
2.2 Genes associated with Ischemic stroke
We have also collected a set of genes which are associated with Ischemic stroke and other types of strokes. These genes list is obtained by performing a detailed literature survey. Majority of the genes are Ischemic stroke associated. Around 48 genes are functionally annotated as stroke associated. A recent survey also listed genes involved in both monogenic and polygenic disorders that may lead to stroke (Ekkert et al., 2022).
2.3 Single nucleotide polymorphism and structural variants from whole genome of Saudis
Filtered and phased genotype data which are biallelic genotype derived from haplotype of each chromosome was analyzed. We merged samples from the two groups SaudiA and SaudiB into a single variant pool. Common and unique variants from both samples using bcftools merge (Li, 2011). We keep the same filtration criteria after merging the two group of samples, which involves k-mer size of 10 to 25, minimum number of reads per region as 10, maximum number of reads in a region being 10000, minimum quality score of 20. Around 4.5 million single nucleotide polymorphisms are obtained from each sample. Besides these insertions/deletions, which are of size range 10 bp to 10 kb is also observed in smaller proportion. Further analysis was focused on the single nucleotide polymorphisms only leaving aside the smaller INDELS.
2.4 Annotation of merged variants using Annovar, dbSNP and RefGene
Merging the SNPs resulted in 4,146,353 unique variants. We indexed dbSNP150 with allelic splitting and left-normalization to annotate these SNPs dbSNP (Sherry et al., 2001). Filter-based annotation module in ANNOVAR pipeline was used to scan the known variants (Wang et al., 2010). Total number of bins analyzed was 28304406. Further, Gene-annotation module was ultilized to annotate variants against hg38 RefGene collection (O'Leary et al., 2016). Around 28,307 genes and Cytoband information was added to the database.
2.5 Functional prediction of variants by comparing the variants to whole-exome data
Functional prediction and annotation of all potential non-synonymous single-nucleotide variants (nsSNVs) in the human genome was performed using dbNSF4 (Liu et al., 2016). This includes a collection of metrics such as SIFT scores, PolyPhen2 HDIV scores, PolyPhen2 HVAR scores, LRT scores, MutationTaster scores, MutationAssessor score, FATHMM scores, GERP++ scores, PhyloP scores and SiPhy scores in version dbnsfp42c.
2.6 Disease specific variants and protein change
Variations which are linked to human health is aggregated in Clinivar database (Landrum et al., 2014). Variant which are of clinical Significance, including pathogenic, non-pathogenic and drug-response, histocompatibility is added to the annotation through ANNOVAR. Prediction of effects of protein-coding variants at the structural and molecular level was performed using SNPeff (Cingolani et al., 2012).
3 Results
We have retrieved 29 samples of two groups of Saudi individuals from 137 whole genome sequencing from Amari et al. Depth and coverage of these samples remain almost similar (Fig. 1).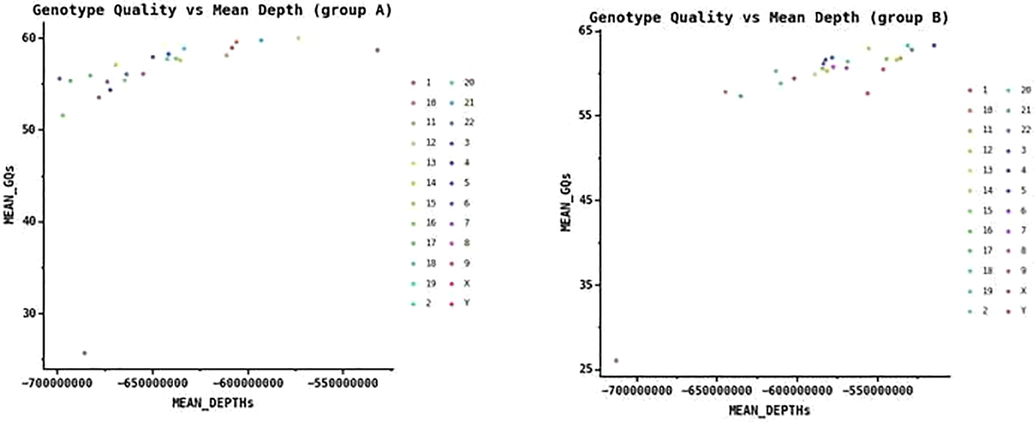
Scatterplot showing the sequencing depth and sequencing quality of the SNPs. (a) sequencing depth and sequencing quality of the SNPs of individual from groupA population (b) sequencing depth and sequencing quality of the SNPs of individual from groupB population.
3.1 Analysis of genetic variation in whole genome of Saudi population
We merged variants from two groups of individuals of Saudi population which resulted in 4,146,365 unique variants. Binning of these variants on individual chromosome and visualization of density histogram on a genome browser show (Fig. 2). These variants are evenly distributed on all chromosomes in both individuals meaning that there is genomic variation similarity (Fig. 3a and b).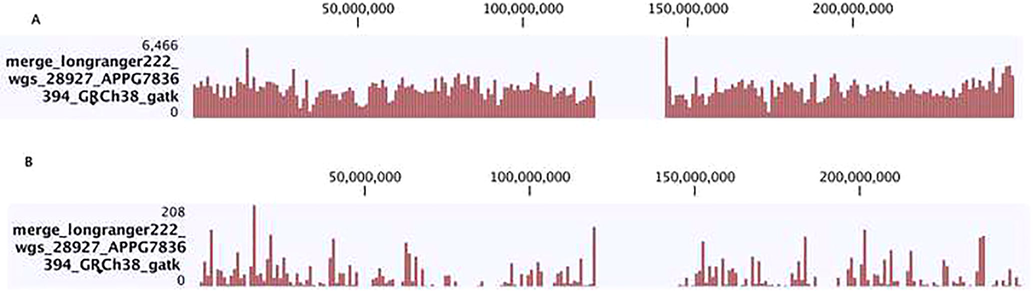
Genome browser view of SNP distribution over 10 kb bins. (a) SNP density before filtering (b) SNP density distribution after filtering and overlapping over Refseq genes collection.
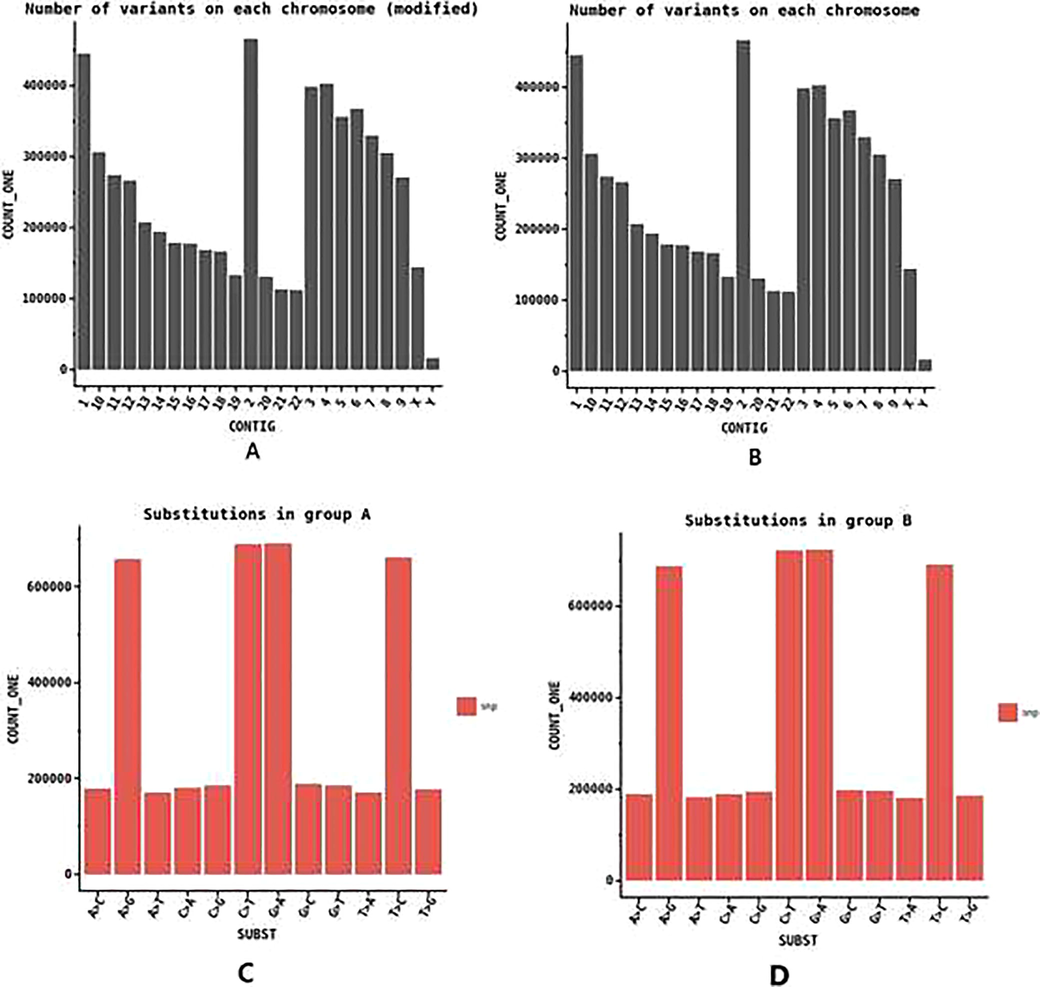
Single nucleotide polymorphism counts over each chromosome. (a) Chromosome wise distribution of genetic variation in groupA Saudi population; (b) Chromosome wise distribution of genetic variation in groupB Saudi population; (c) Barplot showing the count of different types of nucleotide substitution observed in groupA Saudi population and (d) Type of nucleotide substitution in groupB Saudi population.
3.2 Distribution of substitution type and bias in variation
Distribution of different substitution types for the variants, which passed the filter criteria shows a specific pattern. Most observed SNP conversions are G > A and C > T, which is around 35% of the total variant derived from the substitutions in both groups. Other substitutions such A > C, C > G etc are equally low abundant. A > T and T > A are the least abundant of all substitutions (Fig. 3c and d). Homozygous SNPs which have the same allele on both homologous chromosomes of the non-reference genome, and which is different from the reference genome is almost equally represented as that of heterozygous SNPs (Fig. 4a and b).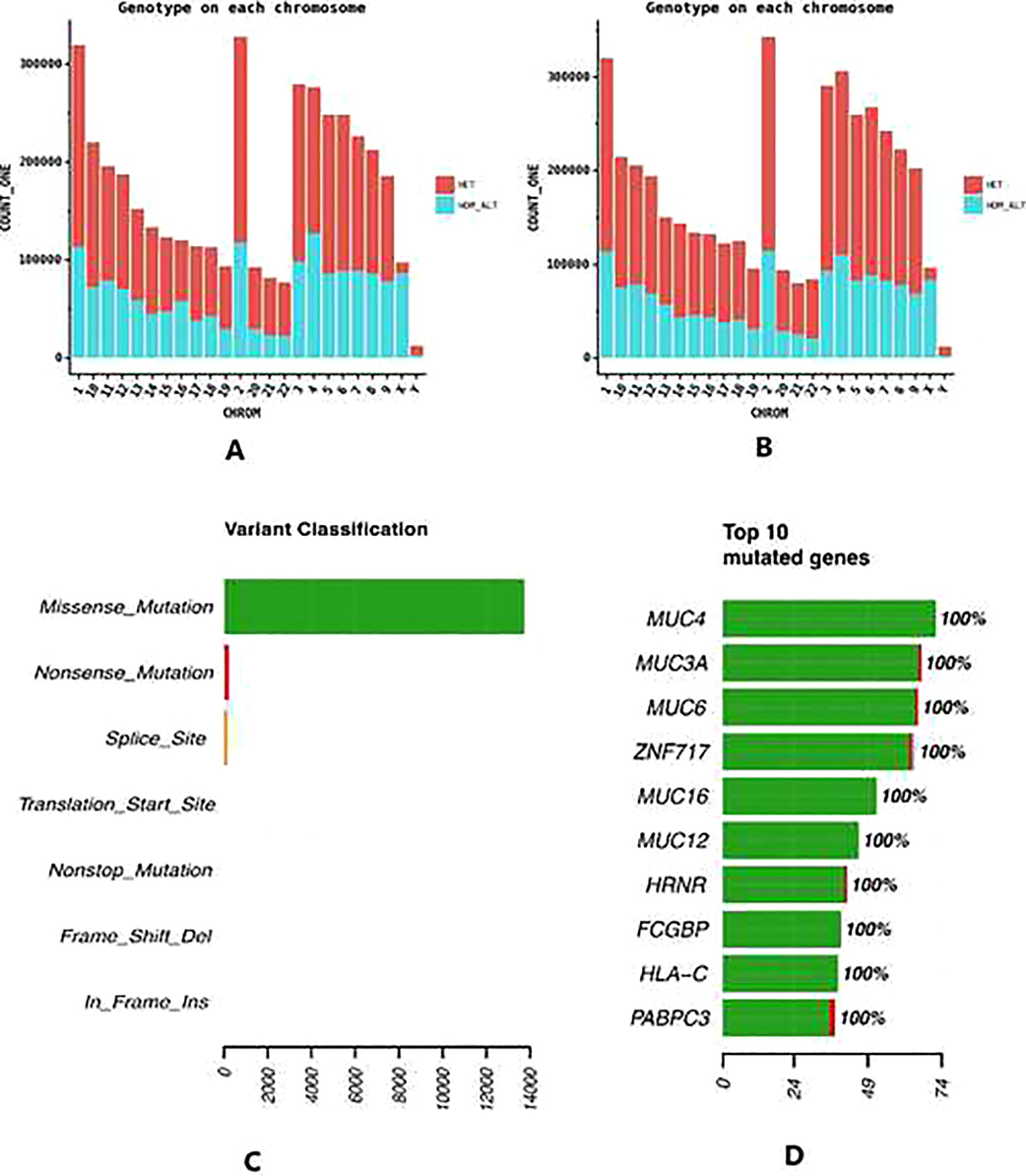
Stacked barplot showing the homozygous SNPs and heterozygous SNPs (a) Allelic variations in groupA Saudi population; (b) Allelic variations in group B Saudi population; (c) Barplot of count of different categories of SNPs based on whole exome based functional annotation using dbNSF4; (d) Top 10 genes harboring most genetic variation resulting from Annovar based annotation of genetic variation using RefSeq database.
We determined the global nucleotide mismatch rate profile for sequencing reads in each tumor sample across all 6 nt substitution types; A·T > C·G, A·T > G·C, A·T > T·A, C·G > A·T, C·G > G·C and C·G > T·A.
3.3 Annotations of mutations that are deleterious or malignant
Among the total number of 4,146,365 SNPs merged, around 10% which is 322,183 SNPs are not observed in dbSNP. ANNOVAR based scanning of avsnp150 database resulted in filtering out 3,824,182 SNPs. Saudi genome specific catalog of known variants and pathogenic variants in was constructed. This is constructed in comparison to the human genome version GRch38. ClinVar database at NIH maintains archives of human genetic variants and annotation with interpretations of their significance to disease. Scanning ClinVar version clinvar_20210501 results in 12,698 SNPs being classified as Benign, 103 SNPs as pathogenic and 629 SNPs likely benign or response to drug (Table2).
SI
Gene
Functional Annotation
1
FCGBP
Fc gamma binding protein(FCGBP)
2
FLG
filaggrin(FLG)
3
HLA − A
Major Histocompatibility Complex, Class I, A
4
HLA − B
Major Histocompatibility Complex, Class I, B
5
HLA − C
Major Histocompatibility Complex, Class I, C
6
HLA − DRB1
Major Histocompatibility Complex, Class II, DR Beta 1
7
HRNR
hornerin(HRNR)
8
KIR2DL1
killer cell immunoglobulin like receptor, two Ig domains and long cytoplasmic tail 1(KIR2DL1)
9
LILRA2
leukocyte immunoglobulin like receptor A2(LILRA2)
10
LILRB1
leukocyte immunoglobulin like receptor B1(LILRB1)
11
MUC12
mucin 12, cell surface associated(MUC12)
12
MUC16
mucin 16, cell surface associated(MUC16)
13
MUC3A
mucin 3A, cell surface associated(MUC3A)
14
MUC4
mucin 4, cell surface associated(MUC4)
15
MUC5B
mucin 5B, oligomeric mucus/gel-forming(MUC5B)
16
MUC6
mucin 6, oligomeric mucus/gel-forming(MUC6)
17
OR8U1
olfactory receptor family 8 subfamily U member 1(OR8U1)
18
PABPC3
poly(A) binding protein cytoplasmic 3(PABPC3)
19
TAS2R19
taste 2 receptor member 19(TAS2R19)
20
ZNF717
zinc finger protein 717(ZNF717)
3.4 Genes associated with variants
Genomic regions which contain variation on genes, in particular protein coding genes can be associated to several genetic disorders. By scanning and annotating against dbNSF (dbnsf42c), we observed 14,847 variations on whole-exome sequencing collections and 4,131,518 SNPs been dropped (Fig. 4c). We also observed that few of the genes harbor large number of variations (Fig. 4d). These include mucin genes which are high molecular weight glycoproteins involved various functions ranging from maintaining lubrication to cell signaling. For example, mucin 4 (MUC4) is a polymorphic mucin protein involved in multiple functions with gene ontology categories being inflammatory response, cell death, cell motion and homeostatic processes, which are common to many diseases (Chaturvedi et al., 2008). Further, this gene is very large with multiple transcripts and comprising atleast 25 exons. This observation of large number of variants on mucin proteins are expected and we further explored variation on genes involved in stroke.
3.5 Single nucleotide polymorphisms in stroke-associated genes
Both monogenic disorders and polygenic disorders involving genetic variation can lead to stroke. These variants show patterns of Mendelian inheritance. Genes associated with stroke include protein coding ones such as SUPT3H/CDC5L, TSPAN2, NOTCH3, TREX1, HTRA1, GLA, NOS3, F5, AALOX5AP, PRKCH, HDAC9, CDKN2A/CDKN2B, EDNRA, TM4SF4-TM4Sn, LINC01492, PITX2, ZFHX3, ZNF566, PDZK1IP1, RGS7, NKX2-5, HABP2, ADCY2, ANK2, FGA, LOC100505841, CDK6, PDE3A, FURIN-FES, PRPF8, ILF3-SLC44A2, ABO, MMP12, SH2B3, CASZ1, WNT2B, KCNK3, SLC22A7-ZNF318, chr9p21, SH3PXD2A, TBX3, LRCH1, SMARCA4-LDLR, PMF1-SEMA4A, FOXF2, ZCCHC14, COL4A1/COL4A2 and PDE4D. We analyzed these 49 genes for additional mutations. Among this, atleast 13 genes show mutation with amino acid changes in one to multiple positions (Fig. 5). For example, COL4A2 shows missense mutation at 517 and 718 with conversion being R517K and P718S. Similarly, in ZFHX3 two variation which are missense mutation at two different positions, one at 460 and other at 777 resulting in amino acid change of E460Q and V777A.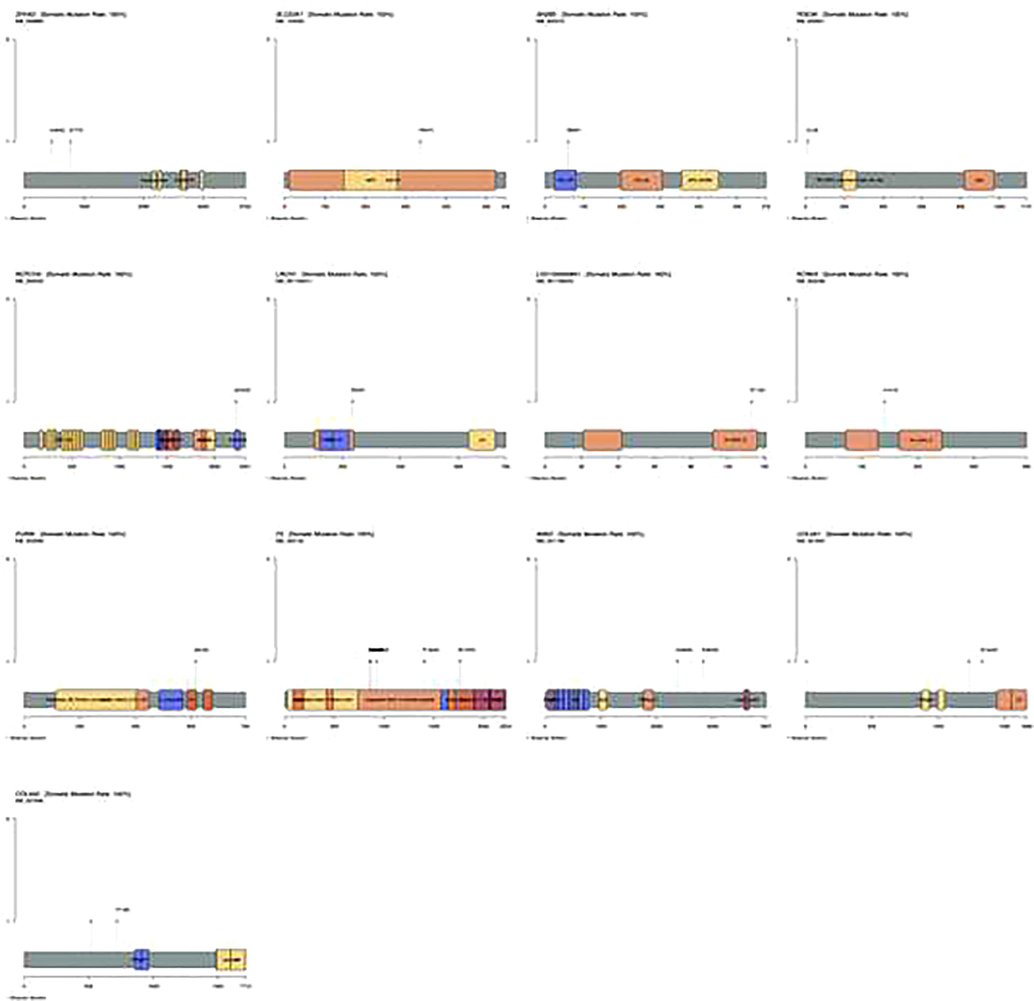
Lollipop plot of genes showing SNPs that could alter the function of the resulting protein by changing the aminoacid (missense mutation).
4 Discussion
Analysis of genome sequencing data from 29 samples of two groups of Saudi individuals from 137 middle eastern populations with around 4.2 million single nucleotide polymorphisms (SNPs) shows that there are large number of genomic variations specific to middle eastern populations. Also, this implies that the available data on genomic variation could be improved by sequencing of human genome of underrepresented ancestry (Scott et al., 2016). Diversifying the genomic data of general population and increasing in cohort size is important for further exploring rare diseases in population of middle eastern ancestry (Abou Tayoun and Rehm, 2020). Substitutions such as G > A and C > T, which is around 35% of the total variant derived from the substitutions in both groups. Other substitutions such A > C, C > G etc are equally low abundant. A > T and T > A are the least abundant of all substitutions. This is observed earlier in other SNP studies reported earlier that the most common change was C > T (or G/A) which is about 70.2%. This may arise from the deamination of 5-methylcytosine at CpG dinucleotides (Dawson et al., 2001).
Genetic contribution to Ischemic stroke risk have been well studied (Rubattu et al., 1996). Ischemic stroke is a manifestation of both monogenic and polygenic disorders with variants on genes being shared among different ancestry. Candidate gene variants detected earlier which are increase in risk of stroke are shared among European and Non-european population, including Chinese population (Ariyaratnam et al., 2007). Our literature survey with resulted in atleast 49 genes involved in risk of stroke. The proportion of strokes of undetermined or rare causes is much higher for young adults and in many cases underlying causes are genetic-related (Jood et al., 2005). We observed that SNPs are extensively distributed in genes which are large and involved in multiple functions. Genome-wide studies on variation on genes also show that these genes are involved in other diseases such as parkinsonian syndrome (Lang et al., 2019).
Variation in genome explains the differences among population such as blood group and susceptibility to diseases (De Mattos, 2013). Monogenic (variation in single gene) or polygenic disorders could be associated to splice-altering variants on protein-coding gene(s) (Vuckovic et al., 2020). Genome sequencing, variant detection and annotation of genotypes are crucial steps in the evaluating, predicting, and concluding diseases in humans (Ku et al., 2010). For example, heterozygous variation in chr17 at position 44,352,876 that we identified in Saudi population is annotated in ClinVar as directly involved in temporal lobar degeneration resulting in susceptibility to Ischemic stroke. In addition, annotation and classification of stroke associated mutation profile and the clusters with similar mutation profiles would help in identifying new subtypes of stroke. Based on this specific treatment strategy differs. Our work contributes to understanding the genetics of stroke and predicts the genomic loci susceptible to stroke. Further work is required for individuals who are susceptible to stroke with a therapy-selection in the future. The result of this study on annotation of variants against clinical database is a valuable contribution as precision medicine based on genome of an individual is the future of health care in Saudi Arabia.
5 Conclusion
A fundamental aspect of this project is to explore the genomic data to enlighten the stroke research using interdisciplinary exploratory approach. Whole genome sequencing and annotation of variation of population of Gulf region including Kuwait, Qatar, and Saudi Arabia expand our understanding of human genetics and diseases in general. Integrating genome sequencing and computational approaches could provide the ability to predict the on-set of stroke and to make possible an early intervention.
Acknowledgements
The authors extend their appreciations to the deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project number (lFP-2020-38).
References
- Genetic variation in the Middle East—an opportunity to advance the human genetics field. Genome Med.. 2020;12:116.
- [Google Scholar]
- Stroke in a Saudi Arabian National Guard community. Analysis of 500 consecutive cases from a population-based hospital. Stroke. 1993;24(11):1635-1639.
- [Google Scholar]
- Stroke register: experience from the eastern province of Saudi Arabia. Cerebrovasc Dis. 1998;8(2):86-89.
- [Google Scholar]
- Common mitochondrial sequence variants in ischemic stroke. Ann Neurol. 2011;69(3):471-480.
- [Google Scholar]
- Genetics of Ischaemic Stroke among Persons of Non-European Descent: A Meta-Analysis of Eight Genes Involving ∼ 32,500 Individuals. PLOS Med.. 2007;4(4)
- [Google Scholar]
- Genome-wide association study identifies a variant in HDAC9 associated with large vessel ischemic stroke. Nat. Genet.. 2012;44:328-333.
- [Google Scholar]
- Stroke in Arab countries: a systematic literature review. J Neurol Sci. 2009;284(1-2):18-23.
- [Google Scholar]
- Structure, evolution, and biology of the MUC4 mucin. FASEB. J. Off. Publ. Fed. Am. Soc. Exp. Biol.. 2008;22:966-981.
- [Google Scholar]
- A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6(2):80-92.
- [Google Scholar]
- A SNP Resource for Human Chromosome 22: Extracting Dense Clusters of SNPs From the Genomic Sequence. Genome Res.. 2001;11(1):170-178.
- [Google Scholar]
- Genetic diversity of the human blood group systems. Rev. Bras. Hematol. E Hemoter.. 2013;35:383-384.
- [Google Scholar]
- Ischemic Stroke Genetics: What Is New and How to Apply It in Clinical Practice? Genes. 2022;13(1):48.
- [Google Scholar]
- Global, regional, and national age-sex-specific mortality and life expectancy, 1950–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392(10159):1684-1735.
- [Google Scholar]
- Family history in ischemic stroke before 70 years of age: the Sahlgrenska Academy Study on Ischemic Stroke. Stroke. 2005;36(7):1383-1387.
- [Google Scholar]
- The discovery of human genetic variations and their use as disease markers: past, present and future. J. Hum. Genet.. 2010;55(7):403-415.
- [Google Scholar]
- ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res.. 2014;42(D1):D980. D985
- [Google Scholar]
- Identification of Shared Genes Between Ischemic Stroke and Parkinson’s Disease Using Genome-Wide Association Studies. Front. Neurol.. 2019;10:297.
- [Google Scholar]
- dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat.. 2016;37(3):235-241.
- [Google Scholar]
- Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res.. 2016;44(D1):D733. D745
- [Google Scholar]
- Stroke in Saudi Arabia: a review of the recent literature. Pan Afr. Med. J.. 2014;17
- [Google Scholar]
- Chromosomal mapping of quantitative trait loci contributing to stroke in a rat model of complex human disease. Nat. Genet.. 1996;13:429-434.
- [Google Scholar]
- Comprehensive gene panels provide advantages over clinical exome sequencing for Mendelian diseases. Genome Biol. 2015;16:134.
- [Google Scholar]
- Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat Genet. 2016;48(9):1071-1076.
- [Google Scholar]
- dbSNP: the NCBI database of genetic variation. Nucleic Acids Res.. 2001;29:308-311.
- [Google Scholar]
- Mendelian and mitochondrial disorders associated with stroke. Semin. Cerebrovasc. Dis. Stroke. 2002;2:46-58.
- [Google Scholar]
- The Polygenic and Monogenic Basis of Blood Traits and Diseases. Cell. 2020;182(5):1214-1231.e11.
- [Google Scholar]
- ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res.. 2010;38(16)
- [Google Scholar]




