

Translate this page into:
Molecular structure, stability and spectroscopic properties of verteporfin and its derivatives – A theoretical insight
⁎Corresponding author. sraju@ksu.edu.sa (Raju Suresh Kumar)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
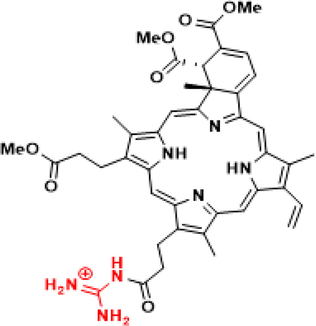
The electronic and geometrical structure of verteporfin VD (1), and its guanidine and dicarboxylic acid derivatives VD(Gua)1 (2), VD(COOH)2 (3), and VD(Gua)3 (4) are analyzed using density functional theory (DFT) calculations. Investigation of compound’s thermal and kinetic stability supports their potential use as photodynamic therapeutics. The results of electronic and spectral analyses by DFT calculation in this work reveal the importance underlying the applications of these compounds.
Abstract
The electronic and geometrical structure of the well-known photosensitizer verteporfin VD (1), as well as its guanidine and dicarboxylic acid derivatives VD(Gua)1 (2), VD(COOH)2 (3), and VD(Gua)3 (4) are analyzed using density functional theory (DFT) calculations. The investigation of compound’s thermal and kinetic stability supports their potential use as photodynamic therapeutics. Though there are reports about the biological applications of verteporfin and its derivatives, the basic structural futures at molecular level which play a crucial role in their potential applications, are not well-explored. The results of electronic and spectral analyses by DFT calculation in this work reveal important points underlying the applications of these compounds.
Keywords
DFT
NMR
PDT
Verteporfin
Guanidine derivative

1 Introduction
Light is essential in the fields of science, the arts and education which cannot be overstated. The healing power of light has been experienced for millennia. Light is a key source in the method known as photodynamic therapy (PDT) which is a photochemical-based treatment. Due to its minimally invasive therapeutic nature, specific attack on the targeted cells, and simplicity of removal from the body, photodynamic therapy is a highly preferred method for the treatment of cancer. The authors A-G. Niculescu et al., (Niculescu and Grumezescu, 2021) and J. H. Correa et al., (Correia et al., 2021) have reviewed the mechanism and applications of PDT up to date and documented a compendium of research reports of PDT as a potential method to treat cancer and the complete details about PDT in the past two decades (Algorri et al., 2021; El-Hussein et al., 2021; Wang et al., 2021; Osuchowski et al., 2021). Also, the interesting facts about the history of PDT are reviewed by G. Gunaydin et al., (Gunaydin et al., 2021). The light sensitive compounds, photosensitizers are important molecules used in photodynamic therapy (PDT). These are categorized as first-generation, second-generation, and third-generation photosensitizers based on their development over time (Niculescu and Grumezescu, 2021). The design and synthesis of innovative photosensitizers for enhanced PDT is an active area of research. Recently, the effectiveness of PDT methods has been improved by the use of nanotechnology for the delivery and action of the photosensitizer (Qiu et al., 2023; Zhou et al., 2023). Continuing from our earlier work (Mahalingam et al., 2018), in which we modified mono guanidine, bisguanidino verteporfin, and triphenyl phosphine-verteporfin derivative for the purpose of effective photosensitization with effective attack on mitochondria, we are now interested in investigating the structural preference that governs the property of Vertiporfin and its derivatives have been reported to have significant biological applications, however thorough structural studies of these compounds at the molecular level are scarce. The mitochondria-targeting photosensitizers visudyne (Mahalingam et al., 2018; Mae et al., 2020; Iacono et al., 2020; Van Dijk et al., 2020), foscon (Chen et al., 2000), and photoferin (Wilson et al., 1997) have already received clinical approval. The key players in the development of efficient photosensitizers are macrocyclic aromatic rings with appropriate substitution. The triphenylphosphine substituted porphyrine derivatives are an important family of photosensitizers with good efficacy in targeting cancer cells (Sibrian-Vazquez et al., 2008). Guanidine substituted aromatic macrocycles, such as porphyrins, are another significant class of photosensitizers (Jiang et al., 2023). The combined therapies, such as photodynamic therapy (PDT) and photothermal therapy (PTT), photodynamic therapy & chemotherapy, and photodynamic therapy & immunotherapy, are also important and showing more advantages in controlling cancer cells (Önal et al., 2022; Li et al., 2022).
Though the biological applications of these photosensitizers in PDT being well-established, there are no clear reports on the preferences of molecular structural configurations of these molecules. Herein, we have investigated the electronic and geometrical structures of verteporfin and their mono guanidino, bisguanidino dicarboxylic acid derivatives at DFT level to understand the basic chemistry beyond these molecules. Spectroscopic studies on these molecules are also not available so far, therefore the DFT-computed spectroscopic characteristics will shed light on their significance. DFT computations have already proven to be successful in understanding the structural features of existing and experimentally unknown molecules. In order to get better photosensitizers with a suitable structural modification, DFT computations are performed and important results from DFT studies are discussed in different headings like geometry, electronic structure, molecular orbital analysis and spectroscopy, etc. In the present study, the photosensitizer verteporfin VD (1) and its guanidine and dicarboxylic acid derivatives VD(Gua)1 (2), VD(COOH)2 (3), and VD(Gua)3 (4), which have already been reported, are chosen to study the electronic and spectral features by DFT calculation.
2 Experimental section
The photosensitizer verteporfin VD (1) and its guanidine and dicarboxylic acid derivatives VD(Gua)1 (2), VD(COOH)2 (3), and VD(Gua)3 (4), which have already been reported, are subjected for DFT calculation.
2.1 Computational details
Computational chemistry methods are becoming important tools for assisting in the complete structural characterization of compounds and for modelling new compounds (Patel and Ganguly, 2022). All the compounds presented in this work were studied using the following strategy: (i) Geometry optimization; (ii) Frequency calculation (iii) NMR property calculation (iv) Molecular Bonding Analysis (v) conceptual DFT reactivity descriptor analysis. All the calculations at density functional theory level were performed using the ORCA programme developed by F. Neese and co-workers (Neese, 2012). The Vosko-Wilk-Nusair parameterization was used for the local density approximation (LDA) with gradient corrections for exchange (Becke88) and correlation (Perdew86) (Vosko et al., 1980; Becke, 1986; Becke, 1986; Perdew, 1986). The TZVP (triple zeta valance with polarisation function) basis set was used for all the molecules (Weigend and Ahlrichs, 2005). In all the calculations, tightSCF convergence criteria were used. The following frequency calculations were evaluated to study optimized geometries in order to check the obtained geometry is the minima (Reveles and Koster, 2004; Schlegel, 2011). Further, the DFT-optimized geometries were used to calculate the NMR parameters like shielding constants, chemical shifts, etc., with the help of the EPRNMR module available in the ORCA software (Mares and Vaara, 2018). For the calculation of 1H and 13C NMR chemical shift values, Tetramethylsilane (TMS) was utilized as a reference (Becker, 2000; Guzman and Hoye, 2022). The reactivity descriptors like chemical potential (μ), hardness (ƞ), softness (S), and electrophilicity (ω) are computed using the HOMO and LUMO energies with the following expressions.: Chemical potential (μ) = ELUMO + HOMO/2; Hardness (ƞ) = ELUMO – HOMO/2; Softness (S) = 1/ƞ; Electrophilicity (ω) = µ2 /2ƞ; The DFT computed global reactivity descriptors have already proven to be successful in predicting the reactivities of the compounds studied and are being used to describe the reactive sites of these important compounds (Tanis et al., 2019; Oller et al., 2018). The DFT optimized geometries and frontier molecular orbital pictures are taken by the visualization software Chemcraft (Chemcraft, 2023).
3 Results and discussion
Synthetic route of the mitochondria target parent photosensitized verteporfin VD (1) and its guanidine and dicarboxylic acid derivatives VD(Gua)1 (2), VD(COOH)2 (3), and VD(Gua)3 (4) is presented in the Fig. 1.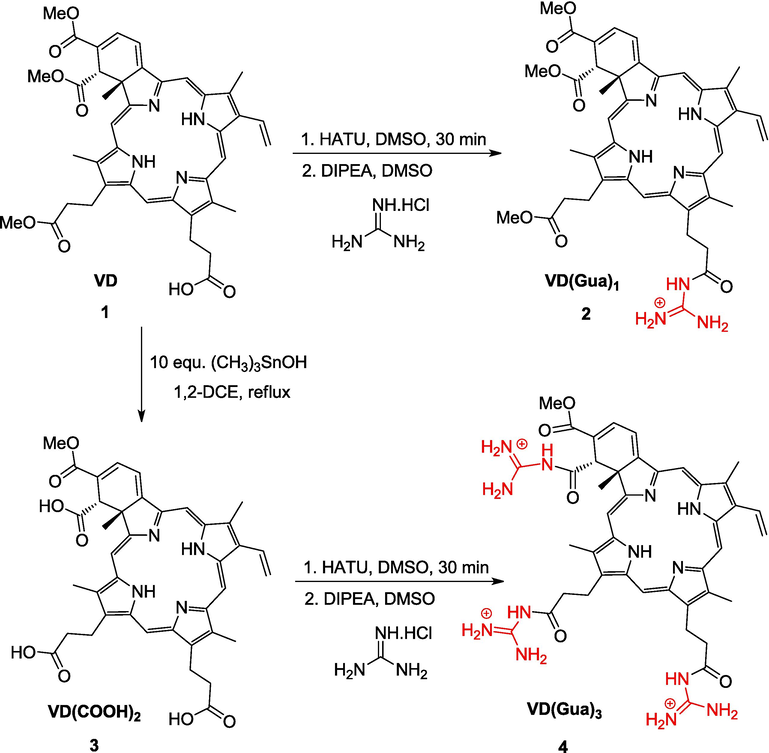
Representation of the modified verteporfin.
Cytotoxicity of the above compounds as photosensitizers has already been well studied. As the structural insights at the molecular level are not well established, the electronic and spectral studies of these compounds were studied by DFT calculations. Computational tools are highly helpful to chemists in studying the structural features of these potential compounds. Modeling of new compounds, designing new strategies for synthesizing novel compounds, and analyzing the existing synthetic routes can be achieved by adopting the proper choice of theoretical methods and analysis tools. The various theoretical aspects like geometrical structure, Reactive descriptor, electronic structure, spectroscopic studies of these photosensitizers are studied and presented in this study.
3.1 Geometrical structure
At the BP86/Def2-TZVP level, the geometry of the VD (1), VD(Gua)1 (2), VD(COOH)2 (3), and VD(Gua)3 (4) compounds is optimized. The DFT optimized geometries of the compound (1–4) are shown in Fig. 2. The N—H bond lengths for all the molecules under study range from 1.02 to 1.022 A0 (1–4). The torsion angle between the four nitrogen atoms in 1 and 3 are 1.45° and 1.44° respectively, indicating that these molecules deviate from the planarity of the porphyrin macrocycle. The torsion angles of the four nitrogen atoms in molecules 2 and 4 are 3.65° and 2.32° respectively, indicating that these molecules deviate more from the planarity of the porphyrin macrocycle. Thus, the substitution of the guanidine moiety increases the loss of planarity of the porphyrin moiety to some extent. Two chiral carbons in the cyclic six-membered ring have angles (C—C—C) of 109.05 and 108.04, indicating that they are sp3 hybridised, while the other angles (C—C—C) of 119.2, 121.67, 118.79, and 120.9, indicating that they are sp2 hybridised. The hydrogen connected to first chiral carbon and the methyl group connected to second chiral carbon of the six membered ring are disposed in trans manner and this trans disposition is confirmed by the H—C*—C*—CH3 angle in compounds 1–4, which ranges from 176.1° to 177°. Though the addition of triphenyl phosphine and guanidine groups in verteporfin affect the anticancer activity of these molecules, from a theoretical perspective, the presence of two chiral carbons in the six-membered ring and the trans disposition of the hydrogen and methyl group bonded to these chiral carbons also play a significant role in determining the biological activity of these compounds.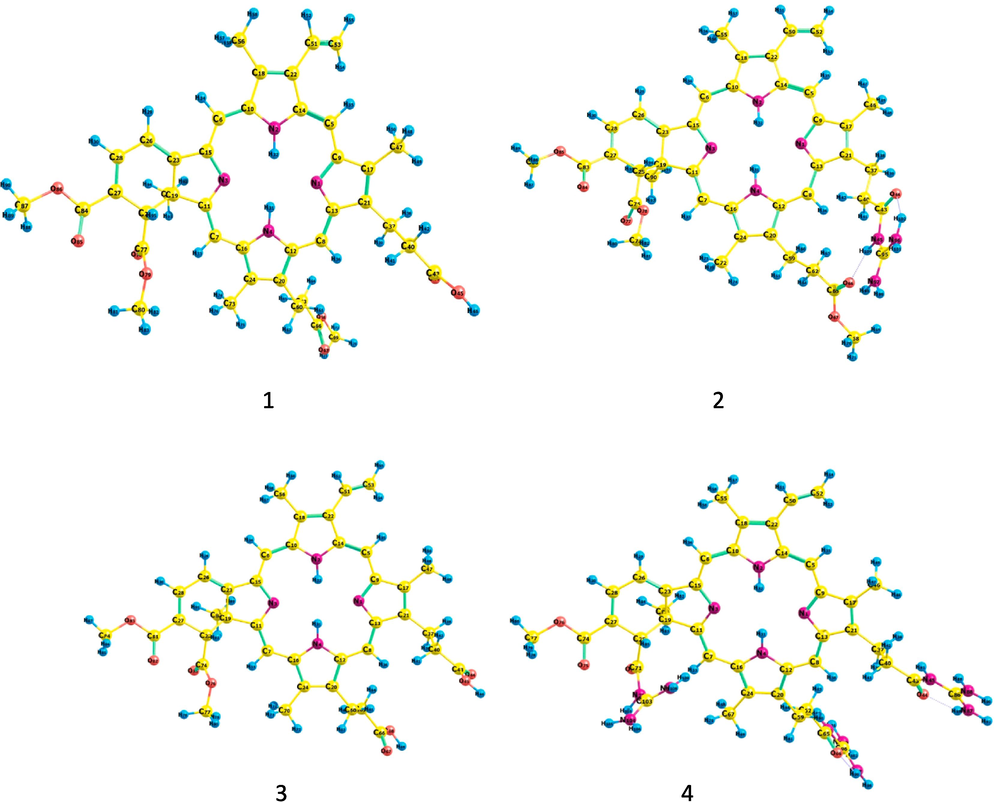
DFT optimized geometries of VD (1), VD(Gua)1 (2), VD(COOH)2 (3) and VD(Gua)3 (4) at BP86/TZVP level.
3.2 Reactive descriptor
Utilizing the ORCA software programme at the BP86/TZVP level, computational studies, i.e., density functional theory (DFT) calculation, are conducted on the compounds VD (1), VD(Gua)1 (2), VD(COOH)2 (3) and VD(Gua)3 (4) (Table 1). The optimized geometries are confirmed to be minima in the potential energy surface by frequency calculations after the optimized geometries are optimized in accordance with the TightSCF. All the four compounds are found to be minima with the considerable differences in energies. The VD(Gua)1 is compared to be more stable than the VD(Gua)3 based on energies. The LUMO-HOMO energy gap has further demonstrated that VD(Gua)1 is more stable than VD(Gua)3 in terms of stability.
Compound
1
2
3
4
HOMO (eV)
−4.8192
−6.4528
−4.8405
−9.9794
LUMO (eV)
−3.3230
−5.5418
−3.3429
−8.9579
E LUMO – HOMO (eV)
1.4962
0.9110
1.4976
1.0215
Chemical potential (µ)
−4.0712
−5.9973
−4.0917
−4.7343
Hardness (□)
0.7481
0.4555
0.7488
0.5108
Softness (S)
1.3367
2.1954
1.3355
1.9577
Electrophilicity (ω)
11.0778
39.4815
11.1792
21.9397
Dipole moment (D)
2.7231
26.4528
3.1482
41.2591
Ionisation potential (eV)
4.8192
4.8405
6.4528
9.9794
Electron Affinity (eV)
3.3230
3.3429
5.5418
8.9579
The geometry optimization of [VD(Gua)3 ie., [C42H50N13O5]3+, converged as local minima with a LUMO-HOMO gap of 1.0 eV, confirming the compound's least stability. The experimental conversion of VD(COOH)2 to VD(Gua)3 was not successful even after many attempts. DFT calculations also supports the least stable character of the compound VD(Gua)3 ie., [C42H50N13O5]3+.
The energy gaps between compounds 1–4 are in the increasing order 3 < 1 < 4 < 2. The reaction between the compound 1 (1.49 eV) and HATU, DMSO, DIPEA, DMSO, and guanidine hydrochloride forms the compound 2, which has a 0.91 eV energy, demonstrating the substance's highly reactive nature. Compound 3 (1.49 eV) does not undergo the reaction and does not form compound 4. However, the computational analysis demonstrates that the compound 4 has possible existence with 1.02 eV energy gap.
Compounds 2 (26.45 Debye) and 4 (41.26 Debye) have very high dipole moments than the remaining compounds 1 and 3 and this high value is attributed to the addition of one and three guanidine groups in compounds 2 and 4 respectively. The higher dipole moment values for 2 and 3 suggest that these can possess better non-linear optical properties.
3.3 Electronic structure
Molecular orbital analysis is a key tool to address the different product formation based on the intrinsic chemical behaviour of each atom involved in bonding. According to the HOMO (highly occupied molecular orbital) energy values, compound 4 requires the highest ionization potential, but compound 1 and 3 can be stable with the lowest ionization values. From the DFT calculations the frontier molecular orbitals are analysed, and the results confirm that VD(Gua)1 is more stable than VD(Gua)3 due to more delocalization of electrons throughout the molecule in VD(Gua)1. For all four of the investigated compounds, the core porphyrin ring and the six-membered ring are the main sources of the HOMO of compound 1. But the electronic contribution from LUMO is differ strongly for compound 2 and 4 when compared to 1, 3. The arrangement of electron delocalization contribute towards the stable nature of 1 and 3 compounds, whereas the LUMO of compounds 2 and 4 are mainly from one of the guanidine group. In compound 4, the LUMO + 1 and LUMO + 2 come from two additional guanidine groups.
Fig. 3 shows the electronic structure of compounds VD (1), VD(Gua)1 (2), VD(COOH)2 (3) and VD(Gua)3 (4). The contribution of HOMO (highly occupied molecular orbital) is highly from the phorphine skeleton at the energy levels of −4.8192, −6.4528, −4.8405, and −9.9794 eV for compound (1–4). In addition, the LUMO (lowest unoccupied molecular orbital) contribution is from substituted groups and marginally in porphine at energy levels of −3.3230, −5.5418, −3.3429, and −8.9579 eV for compounds 1 and 2.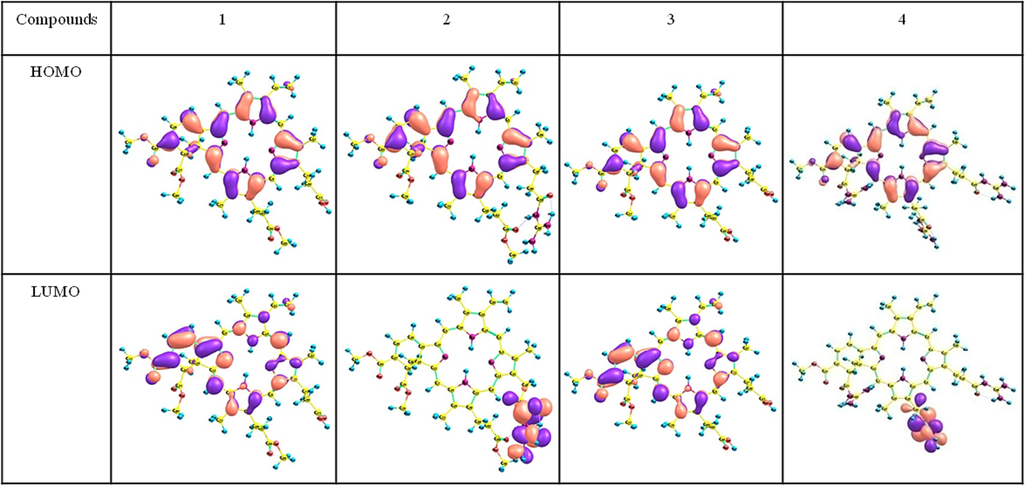
DFT optimized frontier molecular orbitals of compound VD (1), VD(Gua)1 (2), VD(COOH)2 (3) and VD(Gua)3 (4) at BP86/TZVP level.
3.4 Bond order analysis
Mayer bond order analysis is being used as a successful tool in many cases to study the strength of the bonding through bonding analysis (Bridgeman et al., 2001). The Mayer bond order which is a natural extension of Wiberg bond order, has been tested for many inorganic systems containing N-S bond, halogen-oxide molecules and transition metal dichlorides. Here we have used this Mayer bond order analysis to study the strength of the N-C, N–H bonds present in the porphyrin ring and in guanidine moiety. The Mayers Bond order are presented in the Table 2 and Fig. 4.
Atoms
1
2
3
4
N1-C9
1.29
1.29
1.28
1.31
N1-C13
1.29
1.29
1.29
1.27
N2-C10
1.19
1.19
1.20
1.19
N2-C14
1.19
1.19
1.19
1.18
N2-H32
0.87
0.87
0.87
0.88
N3-C11
1.36
1.36
1.33
1.41
N3-C15
1.28
1.28
1.29
1.25
N4-C12
1.18
1.18
1.20
1.18
N4-C16
1.20
1.20
1.19
1.22
N4-H31
0.88
0.88
0.87
0.87
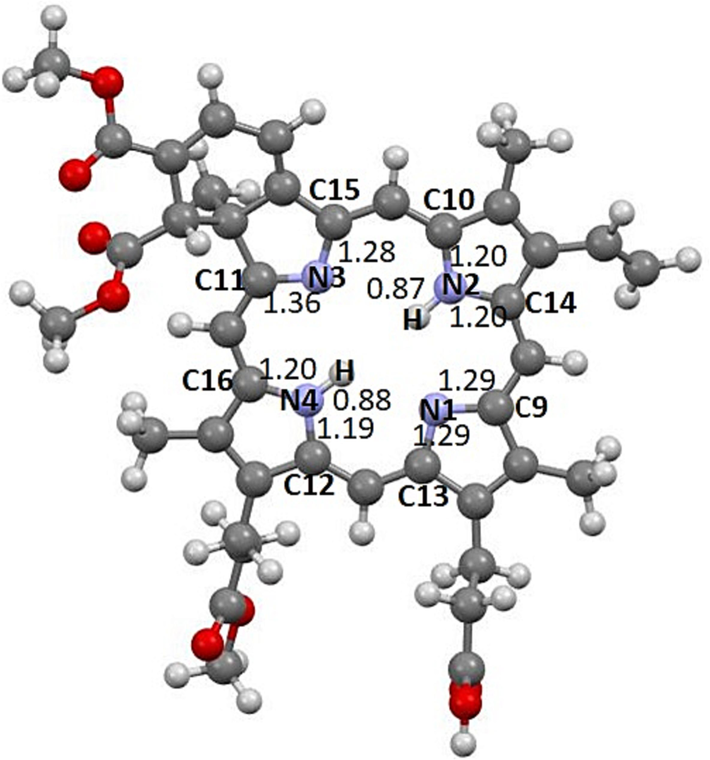
Mayer bond orders of the N-atom connectivity in porphyrine ring in the molecule VD(1).
The Mayer bond order for the compounds VD (1), VD(Gua)1 (2), VD(COOH)2 (3) and VD(Gua)3 (4) calculated at DFT BP86/TZVP level are provided in the Table 2.
It is directly evident that the bonding around the Nitrogen atoms of the central porphyrine ring are strong and their bond order values greater than 1 suggesting the delocalization of the ring electrons which lead to double bond character to the atoms connected to nitrogens except N—H. The two N—H bonds are with the Mayer bond order values 0.87, 0.88 confirm their single bond nature. From the bond order analysis, the highest value of bond order is found in N3-C11 in which C11 is connected to the sp3 carbon. Due to the presence of two -COOMe groups and the two sp3 carbons in connection to C11 which lead to the higher bond order. From the bond order analysis, the C—N bonds of the guanidine moiety shows double bond nature and the N—H bond are single bond as expected.
3.5 Spectroscopic studies
The NMR chemical shift values calculated by DFT are very helpful in understanding these compounds and their uses. The 1H NMR and 13C NMR chemical shift values of VD (1), VD(Gua)1 (2), VD(COOH)2 (3) and VD(Gua)3 (4) are given below.
In 1H NMR, protons H31 and H32 connected to N atoms in molecule VD (1) resonate at −3.20 and −2.99 ppm respectively. Whereas, the N—H hydrogens are resonate at −3.188, −3.00 for (2), at −2.86, −2.86 for (3), and at −2.32, −2.98 ppm for (4), indicating that these particular NH protons are highly shielded. These values are very close to the corresponding value −3.91 ppm of free porphyrine base (Kozlowski et al., 1999). The hydrogens of the C—H groups connecting the four 5-membered indole rings are resonate approximately 9.25, 9.81, 10.31, and 9.77 ppm in VD (1) and the same trend was observed for other three compounds (2), (3) and (4). These chemical shift values are very close to the experimental value 10.43 ppm of the free porphyrin base (Bridgeman et al., 2001).
The nine hydrogens in the three —COOCH3 groups of VD (1) resonate between 3 and 4 ppm, as predicted. The hydrogens of the —COOCH3 groups of compounds (2), (3), and (4) follow a similar pattern. The DFT computation also predicts the chemical shift value of 6.71, 6.36 and 3.82 ppm for the ethylinic hydrogens of compound (1) which is closer to the known ethylinic compounds. The ethylenic groups of compounds (2), (3) and (4) follow the same trend.
The hydrogen of three methyl groups attached to indole ring are showing signal at 3.4–3.6 ppm except the one chemical shift around 2.73, 2.53 and 0.85 ppm which is also connected to six membered ring. This unique chiral centre —CH3 group has a different 1H NMR chemical shift value than the other three molecules. Chemical shift values for compound (2) are 2.75, 2.54, 0.86 ppm, and similar for compounds (3) and (4). The hydrogen of the chiral carbon of the six membered ring in compound (1) resonates at 4.99 ppm, which is another significant result from the DFT calculation. This signal is similar in the remaining compound (2) and (3). Compound (4) exhibits a chemical shift at 5.49 ppm through a higher shielding than compound (1–3).
The remaining two hydrogen atoms H29, H30 of the six membered ring are resonate around 7.69, 7.96 ppm in compound (1). The main reason of the compound veterporfin's high photosensitive activity may be the hydrogen H95 attached to chiral carbon resonates at 4.99 ppm and similar signal is observed in the remaining three compounds.
In the 13C NMR spectrum of compound (1), the —CH- group which connects the indole rings are resonates at 103.20, 94.60, 97.42 and 99.67 ppm for C5, C6, C7, C8 atoms respectively these values are very close to the experimental value of 106.03 ppm observed for the free porphyrin base. Carbon atoms C9, C10, C11 and C12 of each indole ring are resonates at 158.48, 173.22, 138.58, 138.08 ppm, and the expected experimental values are 147, 147, 138.58, and 138.08 ppm, respectively. The carbonyl carbon atom of ester group resonates at 172.42 ppm and the methyl group attached with them is resonates at 55.76 ppm. At 13.11 ppm, the methyl group attached to the indole group resonates. The resonances of the vinyl carbon atoms are 137.05 and 124.40 ppm. At 26.04 and 40.06 ppm, the —CH2- group resonates. The similar trend is followed for compound (2–4). The six-membered acyclic ring's chiral carbon atom exactly resonates at 57.18, 57.52, 57.16, and 56.59 ppm for all the compounds (1), (2), (3), and (4).
4 Conclusions
The verteporfin and its guanidine and dicarboxylic acid derivatives were subjected to DFT(BP86-Def2-TZVP) computations, which provided a clear knowledge of the compounds for their photodynamic therapeutic applications. The DFT optimized geometries are global minima in the potential energy surface and show their possible existence. The computations also predict the more stable nature of the compound 1 and 3 when compared to the stabilities of 2 and 4. Experimental attempts to synthesize compound 4 have failed, and computations also support the the high reactive nature of the modelled triguanidino derivative of the verteporfin 4. DFT calculations estimate the chemical shift values for the individual atoms and the results are extremely similar to those of related compounds or free porphyrin base. The more stable character of compounds 1 and 3 is evident from the frontier molecular orbital analysis revealed by the delocalization of the electrons around the ring in the HOMO and LUMO. The LUMO-HOMO energy gap further confirms the fact that compounds 1 and 3 are more stable than 2 and 4, which are labile. DFT calculations based on the dipole moment values of compounds 2 and 4 show that these compounds may have better non-linear optical properties. The chemical shift values of the individual atoms are determined via theoretical 1H and 13C NMR calculations, which further confirm that the NH protons are highly shielded.
Acknowledgments
The authors extend their appreciation to the Deputyship for Research & Innovation, “Ministry of Education” in Saudi Arabia for funding this research work through the project number (IFKSUDR_E114).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Photodynamic therapy: a compendium of latest reviews. Cancers. 2021;13:4447.
- [CrossRef] [Google Scholar]
- Density functional calculations of molecular bond energies. J. Chem. Phys.. 1986;84:4524-4529.
- [CrossRef] [Google Scholar]
- Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A. 1986;38:3098-3100.
- [CrossRef] [Google Scholar]
- Becker E.D., 2000. High Resolution NMR, third ed. Theory and Chemical Applications, pp. 83-117.
- The Mayer bond order as a tool in inorganic chemistry. J. Chem. Soc., Dalton Trans. 2001:2095-2108.
- [Google Scholar]
- Chemcraft - Graphical program for visualization of quantum chemistry computations (chemcraftprog.com). Available online: https://www.chemcraftprog.com/. Accessed on 1st February 2023.
- The binding characteristics and intracellular localization of temoporfin (mTHPC) in myeloid leukemia cells: phototoxicity and mitochondrial damage. Photochem. Photobiol.. 2000;72:541-547.
- [CrossRef] [Google Scholar]
- Photodynamic therapy review: principles, photosensitizers, applications, and future directions. Pharmaceutics. 2021;13:1332.
- [CrossRef] [Google Scholar]
- A review of chemotherapy and photodynamic therapy for lung cancer treatment. Anti-Cancer Agents Med. Chem.. 2021;21:149-161.
- [CrossRef] [Google Scholar]
- Photodynamic therapy for the treatment and diagnosis of cancer–A review of the current clinical status. Front. Chem.. 2021;9:686303
- [CrossRef] [Google Scholar]
- TMS is superior to residual CHCl3 for use as the internal reference for routine 1H NMR spectra recorded in CDCl3. J. Org. Chem. 2022;87:905-909.
- [CrossRef] [Google Scholar]
- Photodynamic therapy with verteporfin for chronic central serous chorioretinopathy: A review of data and efficacy. Pharmaceuticals. 2020;13:349.
- [CrossRef] [Google Scholar]
- 2D phthalocyanine-assembled porous nanostructure-based electrochemical platform for high-efficiency detection of ascorbic acid. Langmuir. 2023;39:2080-2088.
- [CrossRef] [Google Scholar]
- GIAO nuclear magnetic shielding tensors in free base porphyrin and in magnesium and zinc metalloporphyrins. J. Phys. Chem. A. 1999;103:420-425.
- [CrossRef] [Google Scholar]
- Zein-based nanomedicines for synergistic chemodynamic/photodynamic therapy. ACS Omega. 2022;7:29256-29265.
- [CrossRef] [Google Scholar]
- Verteporfin-photodynamic therapy is effective on gastric cancer cells. Mol. Clin. Oncol.. 2020;13:10.
- [CrossRef] [Google Scholar]
- Targeting of a photosensitizer to the mitochondrion enhances the potency of photodynamic therapy. ACS Omega. 2018;3:6066-6074.
- [CrossRef] [Google Scholar]
- Ab initio paramagnetic NMR shifts via point-dipole approximation in a large magnetic-anisotropy Co(ii) complex. Phys. Chem. Chem. Phys. 2018;20:22547.
- [CrossRef] [Google Scholar]
- The ORCA program system. Wiley Interdiscip. Rev.: Comput. Mol. Sci.. 2012;2:73-78.
- [CrossRef] [Google Scholar]
- Global and local reactivity descriptors based on quadratic and linear energy models for α, β-unsaturated organic compounds. Int. J. Quantum. Chem.. 2018;118:25706.
- [CrossRef] [Google Scholar]
- Development of AB3-type novel phthalocyanine and porphyrin photosensitizers conjugated with triphenylphosphonium for higher photodynamic efficacy. ACS Omega. 2022;7(43):39404-39416.
- [CrossRef] [Google Scholar]
- Photodynamic therapy for prostate cancer–A narrative review. Photodiagnosis Photodyn. Ther.. 2021;33:102158
- [CrossRef] [Google Scholar]
- Exploring the metal-free catalytic reduction of CO2 to methanol with saturated adamantane scaffolds of phosphine-borane frustrated Lewis pair: a DFT study. J. Molec. Graphic. Model. 2022;113:108150
- [Google Scholar]
- Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B. 1986;33:8822-8824.
- [CrossRef] [Google Scholar]
- Dual-photosensitizer nanoplatform based on near-infrared excitation orthogonal emission nanomaterials for enhanced photodynamic therapy of tumors. ACS Appl. Bio Mater. 2023
- [CrossRef] [Google Scholar]
- Geometry optimization in density functional methods. J. Comput. Chem. 2004;25:1109-1116.
- [CrossRef] [Google Scholar]
- Mitochondria targeting by guanidine- and biguanidine- porphyrin photosensitizers. Bioconjugate Chem.. 2008;19:705-713.
- [CrossRef] [Google Scholar]
- Synthesis, characterization, computation of global reactivity descriptors and antiproliferative activity of N-(4-nitrophenyl)Acrylamide. Russ. J. Phys. Chem B. 2019;13:49-61.
- [CrossRef] [Google Scholar]
- Photodynamic therapy for chorioretinal diseases: a practical approach. Ophthalmol. Ther.. 2020;9:329-342.
- [CrossRef] [Google Scholar]
- Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can. J. Phys.. 1980;58:1200-1211.
- [CrossRef] [Google Scholar]
- An update in clinical utilization of photodynamic therapy for lung cancer. J. Cancer. 2021;12:1154-1160.
- [CrossRef] [Google Scholar]
- Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005;7:3297-3305.
- [CrossRef] [Google Scholar]
- Subcellular localization of Photofrin and aminolevulinic acid and photodynamic cross-resistance in vitro in radiation-induced fibrosarcoma cells sensitive or resistant to photofrin-mediated photodynamic therapy. Photochem. Photobiol.. 1997;65:166-176.
- [CrossRef] [Google Scholar]
- Biomimetic upconversion nanoplatform synergizes photodynamic therapy and enhanced radiotherapy against tumor metastasis. ACS Appl. Mater. Interfaces 2023
- [CrossRef] [Google Scholar]




