

Translate this page into:
Molecular Modeling and simulation-based identification of inhibitors against new Delhi Metallo-Lactamase 1: Implications for bacterial antibiotic resistance
⁎Corresponding author at: Research and Scientific Studies Unit, College of Nursing and Health Sciences, Jazan University, Jazan-45142, Saudi Arabia. shhaque@jazanu.edu.sa (Shafiul Haque),
⁎⁎Corresponding author. faraz.ahmad@vit.ac.in (Faraz Ahmad),
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Objectives
Bacterial infections expressing New Delhi metallo-lactamase-1 (NDM-1) pose an escalating global threat to healthcare systems. NDM-1 is an enzyme that renders β-lactam antibiotics ineffective, leading to resistance against numerous antibiotics used in clinical practice. Therefore, there is an urgent need to identify and develop a clinically relevant inhibitor for NDM-1.
Methods
Vitas-M laboratory database was screened for small molecules with abilities to bind NDM-1, by generating structure-based pharmacophore hypothesis. Thereafter, molecular docking was performed between NDM-1 and the potential small molecule inhibitors. The outcomes of molecular docking were validated by molecular dynamics simulation and MM-GBSA protocols.
Results
Based upon initial NDM-1-binding characteristics, two ligands (STK115225 and STK107343) were nominated for further analyses for stability and affinity of protein–ligand interactions. Assessment of conformational change parameters indicated that these showed tight and stable binding to the active site pocket of NDM-1 protein. Principal component analysis (PCA) further illustrated that the protein ligand complexes were highly stable. Molecular dynamics simulation along with high numbers of static hydrogen bonds signifies the potency of STK115225 and STK107343 in inhibiting NDM-1. Further, MM-GBSA-based binding free energy maps verified favorable energy changes for the binding of the two small molecules, indicating their abilities for high affinity-binding with NDM-1.
Conclusions
This study has significant implications for addressing antibiotic resistance mediated by NDM-1. The identification of STK115225 and STK107343 as high-affinity binding ligands against NDM-1 provides a strong foundation for developing new therapeutic agents. However, to fully ascertain their clinical relevance, these findings must be validated through in vitro and in vivo experiments. If successful, these inhibitors could restore the efficacy of β-lactam antibiotics and offer a new approach to combat NDM-1 mediated antibiotic resistance, ultimately improving patient outcomes and reducing the global burden of resistant bacterial infections.
Keywords
NDM-1
Vitas-M laboratory
Structure-based pharmacophore
Virtual screening
MM-GBSA

1 Introduction
Emergence of bacterial antibiotic resistance has become a major clinical health concern (Ahmed et al., 2021; Faheem et al., 2013; Muteeb et al., 2017). β-lactamases enzyme is one of the more recently discovered agent in the bacterial arsenal for antibiotic resistance, and can efficiently hydrolyze β-lactam antibiotics such as penicillins, cephalosporins, monobactams, and carbapenems (Wang et al., 2021). β-lactamases function by hydrolyzing the β-lactam ring available in β-lactam antibiotics. The hydrolysis reaction involves the nucleophilic attack on the carbonyl carbon of the β-lactam ring, leading to the opening of the ring and loss of antibiotic activity. The production of β-lactamases by bacteria significantly complicates the treatment of bacterial infections. Infections caused by organisms that produce β-lactamase are linked to higher morbidity and mortality rates, extended hospital stays, and elevated healthcare costs. The presence of β-lactamases, particularly extended-spectrum beta-lactamases (ESBLs) and carbapenemases like NDM-1, in pathogenic bacteria such as Escherichia coli and Klebsiella pneumoniae has led to outbreaks in healthcare settings and community environments.
Protein homology and underlying molecular mechanisms of action can be used to classify β-lactamases, as suggested by Ambler, into 4 different groups (A, B, C & D). Class A β-lactamases are “serine β-lactamases”, which include enzymes like TEM and SHV, often found in Gram-negative bacteria. Class C β-lactamases are also serine-based, commonly referred to as AmpC enzymes, which are chromosomally encoded and inducible. Class D β-lactamases, known as oxacillinases, have a serine residue at their active site and are capable of hydrolyzing oxacillin and other β-lactams. NDM-1 belongs to class B, also known as metallo-β-lactamases (MBLs). Unlike other classes, metallo-β-lactamases (MBLs) need divalent metal ions, usually zinc, for their enzymatic activity and can break down a broad spectrum of β-lactam antibiotics, including carbapenems and cephalosporins (Hall and Barlow, 2005). Furthermore, Bush and Jacoby have classified β-lactamases into three functional groups (1, 2 and 3). Cephalosporinases are in group 1, while penicillinases, oxacillinases, serine-based carbapenemases, and ESBLs are in group 2. Metal ion-requiring carbapenemases are classed into group 3 (Bush and Jacoby, 2010).
NDM-1, belonging to Class B1 (Ambler’s classification) and Group 3a (Bush and Jacoby’s classification), is a very potent β-lactamase which can inactivate β-lactam antibiotics, including cephalosporins, carbapenems and penicillins (Brem et al., 2016)7. Mechanistically, NDM-1 selectively hydrolyzes amide bonds in β-lactam antibiotics. NDM-1 was discovered in a Swedish patient in India in 2008, who was diagnosed with urinary tract infection of Klebsiella pneumoniae (Kumarasamy et al., 2010). Since then, NDM-1 has been detected in numerous countries across all continents, carried by various bacterial species, including Escherichia coli, Klebsiella pneumoniae, Acinetobacter baumanii, and Enterobacter cloacae (Linciano et al., 2019). The gene encoding NDM-1 is often located on plasmids, which are mobile genetic elements that facilitate horizontal gene transfer between bacteria, further accelerating the spread of resistance. This plasmid-mediated transfer allows NDM-1 to disseminate rapidly across different environments, from hospitals to community settings, complicating infection control efforts. The high prevalence of NDM-1 in both clinical and environmental isolates indicates its widespread dissemination and the urgent need for coordinated surveillance and containment strategies.
Due to the serious consequences of NDM-1-mediated antibiotic resistance, it is urgently necessary to find new inhibitors that can effectively target this enzyme. Current treatment options are limited and often involve the use of antibiotics that are more toxic or less effective, such as colistin and tigecycline. These alternatives may have adverse side effects and are not always successful in treating infections caused by NDM-1-producing bacteria. Research efforts are focused on developing new molecules that can inhibit the activity of NDM-1, thereby restoring the efficacy of beta-lactam antibiotics. Strategies include designing small molecules that can chelate the zinc ions essential for NDM-1 activity or developing novel compounds that can bind to the enzyme's active site. While NDM-1 has been proposed to be repressed by chemicals such as captoprils, sulfonamides, boric acid derivatives and thiol-containing small molecules, none of these potential inhibitors have received approval for clinical use. Hence, in order to counteract bacterial antibiotic resistance, it is essential to design/identify, evaluate and further develop novel and potent NDM-1 inhibitors (Klingler et al., 2015).
Computer-assisted drug design (CADD) has revolutionized the field of pharmaceutical research, offering significant advantages in the drug discovery process. However, it also comes with several limitations that need to be addressed for optimal application. These include the complexity and dynamic nature of biological systems, which are often oversimplified in computational models, leading to inaccuracies. The accuracy of predictive models depends on the quality of data, which can be incomplete or biased, and the need for substantial computational resources can be a barrier. Modeling the flexibility of biological targets is challenging, and integrating computational predictions with experimental data is time-consuming and costly. Scalability issues, outdated software and algorithms, and difficulties in accurately predicting ADMET properties further complicate the drug discovery process. Continuous advancements and improved integration with experimental data are needed to fully realize the potential of CADD.
Given the substantial role of NDM-1 in the dissemination of antibiotic resistance, this in silico study is dedicated to identifying potential small molecule inhibitors of NDM-1.
2 Methodology
2.1 Pharmacophore hypothesis
Protein Data Bank (PDB) was used for the retrieval of co-crystal structures of NDM-1 (PDB IDs 4EY2, 4EYB, 4EYL, 4RL0, 6OL8, and 6TTC). The structures were superposed to analyze the orientation of the binding pocket residues. The residues with consistent conformation were then selected for the generation of pharmacophore hypothesis. A pharmacophore hypothesis was developed by selecting the receptor cavity and manually choosing the binding pocket residues, utilizing the Phase tool of Schrödinger (Dixon et al., 2006). Before developing the hypothesis, the receptor was prepared by following the steps outlined in section 2.4.
2.2 Small molecule database creation and virtual screening
Phase tool was also used to create a database of small molecule compounds from the Vitas-M laboratory database, containing 1.4 million ligands (Dixon et al., 2006). Ten conformers were produced for every ligand in order to expand the chemical space search. Using the Epik tool (Shelley et al., 2007), various potential states at pH 7 were produced, and the database was cleared of the high energy tautomeric forms. Pharmacophore hypothesis was then applied to the virtual screening using the prepared database. For the molecular docking investigations, the screened hits with phase scores greater than 1.4 were chosen.
2.3 Molecular docking
Molecular docking of a small molecule database against NDM-1 was conducted using Glide (Schrodinger, LLC, NY, USA). Protein preparation wizard of Maestro was exploited to create the crystal structure of NDM-1 (PDB ID 4EYL) (Madhavi Sastry et al., 2013). Preprocessing of the receptor included adding hydrogens, eliminating water, adding charges, and repairing side chain atom residues. Using PROPKA, the superfluous chains were cut off and tautomeric forms were produced at a pH value of 7.0 (Bibi Sadeer et al., 2019). OPLS_2005 force field was employed to further optimize and minimize the receptor’s shape (Shivakumar et al., 2012). Creation of the grid relied on using the co-crystal ligand to perform site-specific docking. The dimension of the grid box was set as 27 Å × 27 Å × 27 Å, while the X, Y, and Z coordinates were 2.51 Å, −40.8 Å, and 1.89 Å, respectively. Prior to docking, the ligands were synthesized using Maestro’s LigPrep tool following grid creation (Matsuoka et al., 2017). Epik was used to produce ionization states at a set pH value of 7.0 (Shelley et al., 2007). Stereoisomers of the selected ligands with the specific chirality were created employing OPLS_2005 force field tool. Van der Waals radii of receptor atoms were set at 1.0 and the partial charge cut-off value was scaled at 0.25 to minimize the potential of the receptor’s non-polar portions. Subsequently, Glide docking tool was employed to dock the ligands to the synthesized receptor, NDM-1. Binding poses for the ligand-receptor interactions were examined using the glide gscore.
2.4 Molecular dynamics simulations (MDS)
Following preprocessing of the ligand-receptor complexes using OPLS_2005 force field, assessment of dynamic interactions via MDS for the selected protein–ligand complexes obtained using molecular docking was performed at 100 ns on Desmond software of Schrödinger (Bowers et al., 2006). Transferable intermolecular potential with three points (TIP3P) solvent model and an orthorhombic box (10 × 10 × 10 Å) was added using the system builder tool (Price and Brooks, 2004). Counter ions (0.15 M Na+/Cl-) were supplemented in the model to neutralize it and simulate the physiological conditions. Isothermal–isobaric (NPT; amount of substance N, pressure P and temperature T) parameters were set at 300 K and 1 atm. Prior to initiating MDS, the complex was loosened, and every 50 ps, trajectories were saved for the purpose of analyzing the simulation's output.
2.5 Dynamic Cross-Correlation matrix (DCCM)
The Dynamic Cross-Correlation Matrix (DCCM) method is a valuable computational tool used to analyze the correlated motions of atomic pairs within molecular systems, particularly proteins and their interactions with ligands. This method involves conducting molecular dynamics simulations to produce trajectories of atomic coordinates over time. It then eliminates global translational and rotational motions by superimposing these coordinates onto a reference structure. The method calculates the displacement vectors of atoms from their mean positions and constructs a cross-correlation matrix based on the correlation coefficients between these displacement vectors. For each pair of atoms i and j, the displacement vectors δri(t) and δrj(t) from their respective mean positions were computed over the course of the simulation. These vectors represent the deviations from their average positions at any given time t. The DCCM is constructed by calculating the cross-correlation coefficients between the displacement vectors of each pair of atoms. The elements of the DCCM, Cij, are given by the following equation: where 〈⋅〉 denotes the time average over the entire simulation trajectory.
2.6 Principal component analysis (PCA)
Principal Component Analysis (PCA) was used to evaluate the collective motions of both proteins and their accompanying ligands, utilizing the Bio3D package (Grant et al., 2021). The process initiated with the elimination of translational and rotational motions of the protein. Subsequently, the covariance matrix and its corresponding eigenvectors were computed by aligning the atomic coordinates of the protein to a reference structure. This symmetric matrix was then diagonalized via an orthogonal transformation matrix, yielding a diagonal matrix of eigenvalues. The covariance matrix (C) was calculated using the following equation: where, N, xi/j and < xi/j > signify the number of Cα-atom, the Cartesian coordinate of the ith/jth Cα-atom, and the time average of all the conformations, respectively.
2.7 Molecular Mechanics-Generalized born surface area (MM-GBSA)
The free energies for the interaction between NDM-1 and the selected ligands were computed using molecular mechanics force fields and the implicit solvation method using Prime (Schrodinger, LLC, NY, USA), as previously described (Ahmed et al., 2020; Friesner et al., 2006). Initially, the energies of the docked poses were minimized using Prime’s local optimization feature. Subsequently, the binding energies were computed using an MM/GBSA continuum solvation protocol, employing the OPLS3a force field, a VSGB solvation model, and a rotamer search algorithm (AlAjmi et al., 2021). The free energy was calculated according to the following relationship:
where GComplex_minimized, GLigand_minimized, and GProtein_minimized were the minimized free energies of the protein–ligand complex, ligand only, and protein only, respectively.
3 Results
3.1 Protein structures alignment
After retrieval of protein crystal structures of NDM-1 from PDB, they were aligned on each other to analyze the binding pocket residues. The RMSD difference was less than 0.5 which showed that there were not many deviations in the structures (Fig. 1A). The orientation of the residues of binding pocket was deeply analyzed (Fig. 1B). It was observed that the conformations of Val73, Trp93, His122, Gln123, Asp124, and Lys211 had changed in 6TTC structure.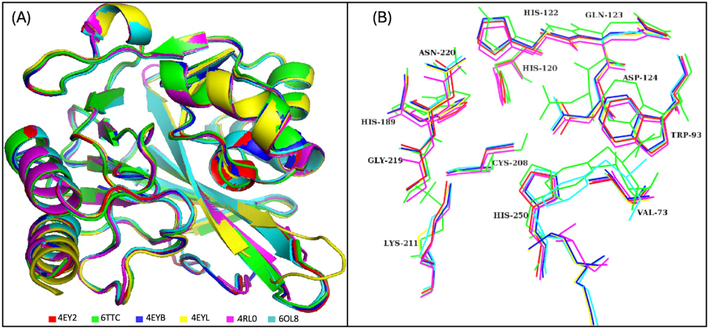
The superimposed residues of the NDM-1 crystal structures used for pharmacophore model generation. (A) The alignment of the co-crystal structures, (B) The aligned residues of the binding pocket.
3.2 Pharmacophore creation
Binding pocket residues of NDM-1 protein were chosen in order to create a seven-featured pharmacophore model of the receptor cavity. The coordinates in the protein structure for the pharmacophore hypothesis are depicted in Fig. 2A and summarized in Sup. Table S1. Fig. 2B depicts the features of the pharmacophore hypothesis, including the binding pocket cavity.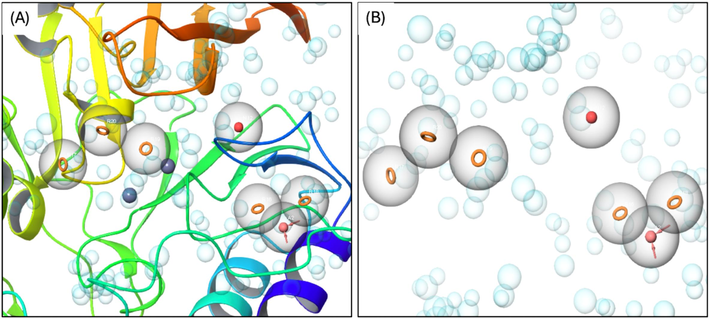
Pharmacophore model of NDM-1 for the binding pocket (A) and the cavity of the pocket (B).
Energy components (kcal/mol)
STK115225-complex
STK107343-complex
ΔEvdW
−52.86 ± 0.34
−59.85 ± 0.37
ΔEele
−2.96 ± 0.25
−8.90 ± 0.36
ΔEGB
13.40 ± 0.22
20.85 ± 0.32
ΔEsurf
−5.50 ± 0.03
−5.63 ± 0.04
ΔGgas
−55.83 ± 0.37
−68.75 ± 0.54
ΔGsolv
7.89 ± 0.21
15.21 ± 0.32
ΔGtotal
−47.93 ± 0.31
−53.54 ± 0.43
3.3 Virtual screening
Virtual screening of the selected ligands was conducted employing the pharmacophore model of NDM-1. A compound was designated as a hit only when a minimum of four features were found to be matched. Parameters such as vector alignments, RMSD site matching and volume scores were used to rank the final hits from screening in order of their phase fitness scores. Selected ligands’ vector scores ranged from −1.0 to + 1.0., and higher values in the vector score corresponded to greater alignment. On the other hand, volume scores ranged between 0.0 and 1.0, with higher volume scores indicating higher commonality of the volumes of the aligned and reference ligands. Phase screen score threshold was set at 1.4 in order to identify possible hits. The ligands which were selected along with their phase screen score and alignment on the pharmacophore hypothesis are given in Sup. Table S2.
S. No.
Compound ID
Structure of ligand
Glide score
1.
STK115225
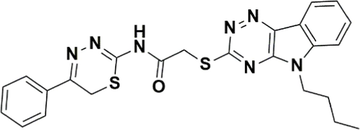
−7.291
2.
STK107343
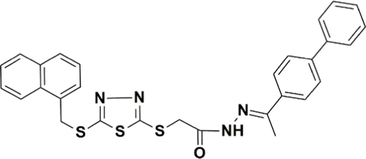
−7.280
3.
STK215880
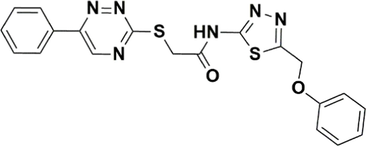
−7.262
4.
STK115575
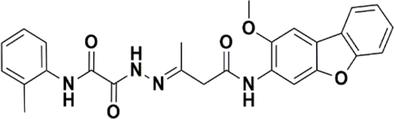
−7.043
3.4 Molecular docking
The ligands were docked to NDM-1 protein in accordance with the standard methodology of the glide tool. Analyses of molecular interactions between the compounds docked to the receptor were evaluated in consideration with the glide g-score obtained. A cutoff binding energy score of −7 kcal/mol was fixed in order to select the hits. This resulted in the selection of 4 hits with appropriate glide g-score and binding energies (Table 2). Upon analyses of the molecular interactions between the protein and docked hits, we observed that STK115225 made one π-π interaction with His122, and four π-alkyl interactions with Leu65, Val73, Trp93, and His250. STK107343 was found to make one π-sulfur bond with Trp93, one π-σ bond with His122, a π-π bond with His250, and an alkyl bond with Ala215. STK215880 was involved in one π-π interaction with His250, an alkyl bond with Ala215 and a metal acceptor with Zn as well. Lastly, STK115575 elicited three hydrophobic interactions with Ala74, Trp93, and His122. Fig. 3 depicts the ligand-receptor molecular interactions for all the four hits against NDM-1. Analyses of binding modes for the selected ligands docked to NDM-1 were also conducted and are represented in Fig. 4.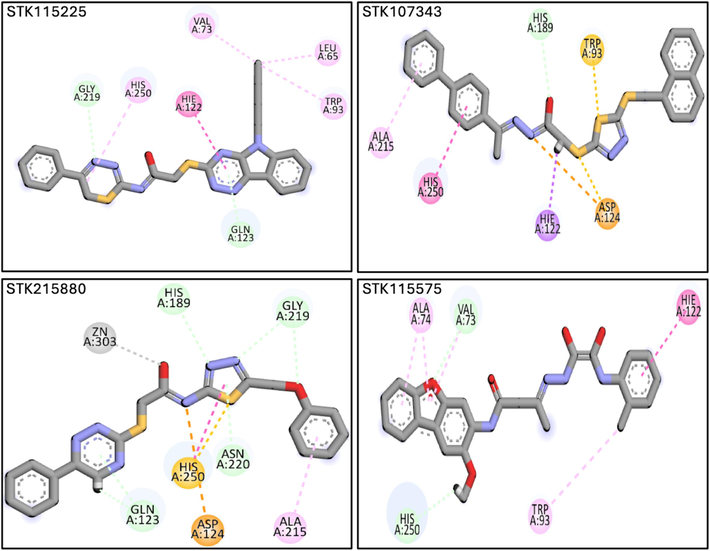
Molecular interactions underlying binding of the four selected ligands (STK115225, STK107343, STK215880 and STK115575) to NDM-1. Magenta, purple and orange lines depict hydrophobic interactions, pi-Sigma bonds, and pi-sulfur interactions, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
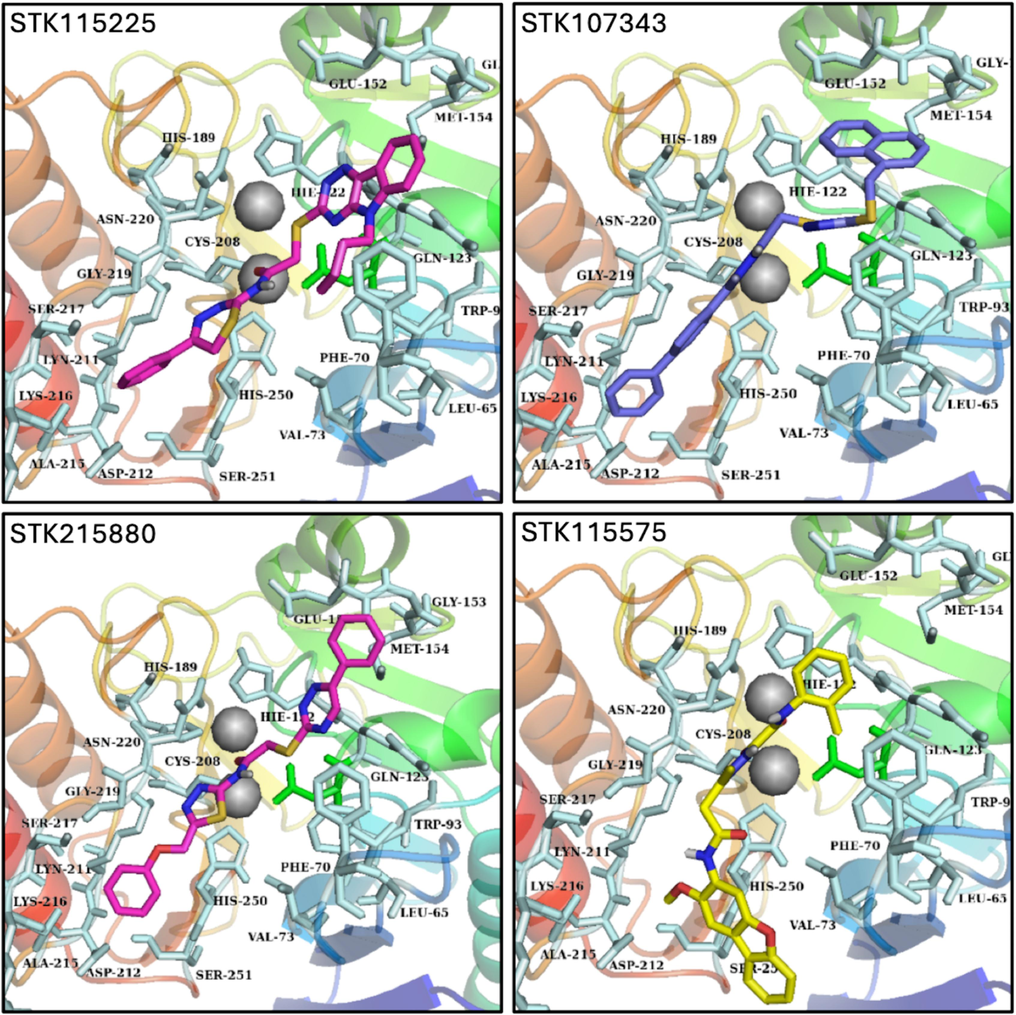
Probable binding modes of the four selected ligands (STK115225, STK107343, STK215880 and STK115575) represented as sticks in the binding pocket of NDM-1.
3.5 MDS analysis
The docked poses of the ligands were superposed on the co-crystal ligand and two compounds were chosen for protein–ligand stability analyses. The aligned poses of the selected compounds are shown in Fig. 5 A-B.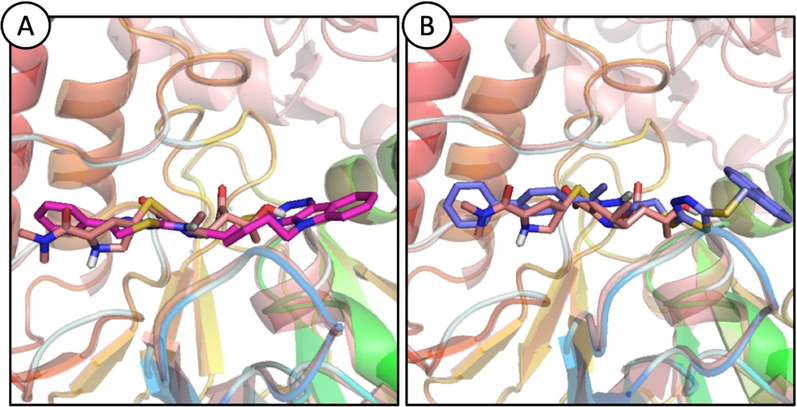
Probable binding modes for ligands, STK115225 (A; magenta sticks) and STK107343 (B; blue sticks) aligned with NDM-1 co-crystal structure. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.5.1 Root mean square deviation (RMSD)
To evaluate the stability of the ligand–protein complexes, RMSD of the alpha carbon atoms in the proteins and ligands was computed from the trajectories (Sargsyan et al., 2017). As depicted in Fig. 6A, complexes for STK115225 and STK107343 and were equilibrated for 10 ns. RMSD of STK115225 complex showed negligible deviations till 20 ns and achieved stability at ∼ 1.25–1.50 Å until the culmination of the MDS. There were minor deviations at the frames generated at 50 and 60 ns. For the STK107343 complex, RMSD attained a value of ∼ 1.75 Å and remained in this range until the culmination of the MDS at 100 ns.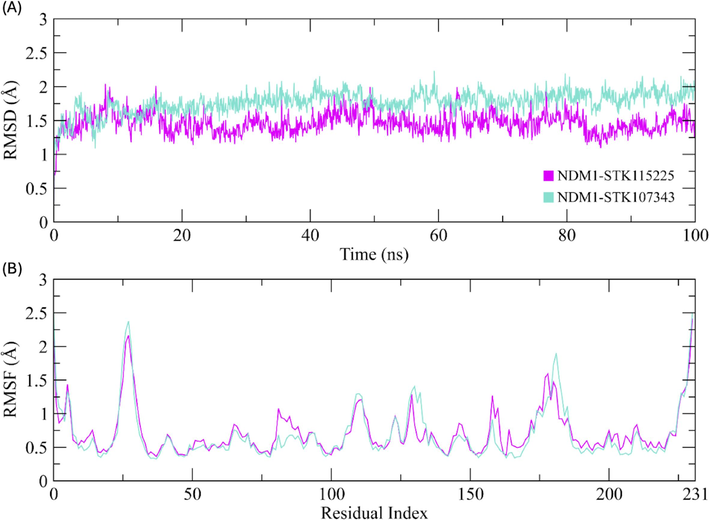
(A) RMSD plots of ligand–protein complex for the duration of MDS (100 ns). (B) RSMF value plots depicting the residue fluctuations during the simulation period. Cyan and magenta plots represent complexes of STK107343 and STK115225, respectively for both (A) and (B). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.5.1.1 Root mean square fluctuations (RMSF)
RMSF values are a good measure of the dynamic behavior of amino acid restudies in receptor proteins, when the latter are complexed with their ligands (Martínez, 2015). With the exception of the loop section residues which extended to the maximum value of ∼ 2.5 Å, RMSF of other residues were found to vary by less than 1 Å during the course of the simulation (Fig. 6B). Overall, the results indicate that the protein residues are more-or-less rigid and do not show any major fluctuations. This suggests that the ligands form a stable complex with the protein receptor DNM1.
3.5.1.2 Protein-Ligand contacts
Based on MDS analysis, interactions between the ligands and receptor protein, NDM-1 mainly relied on hydrophobic, ionic, and hydrogen bond interactions. Amino acid residues involved in ionic bonds in STK115225-NDM-1 complex were Asp124, Cys208, Lys211, and His250 (Fig. 7A). On the other hand, His120, His122, Asp124, His189, Cys208, Lys211 and His250 were found to make ionic bonds with STK107343 compound (Fig. 7B).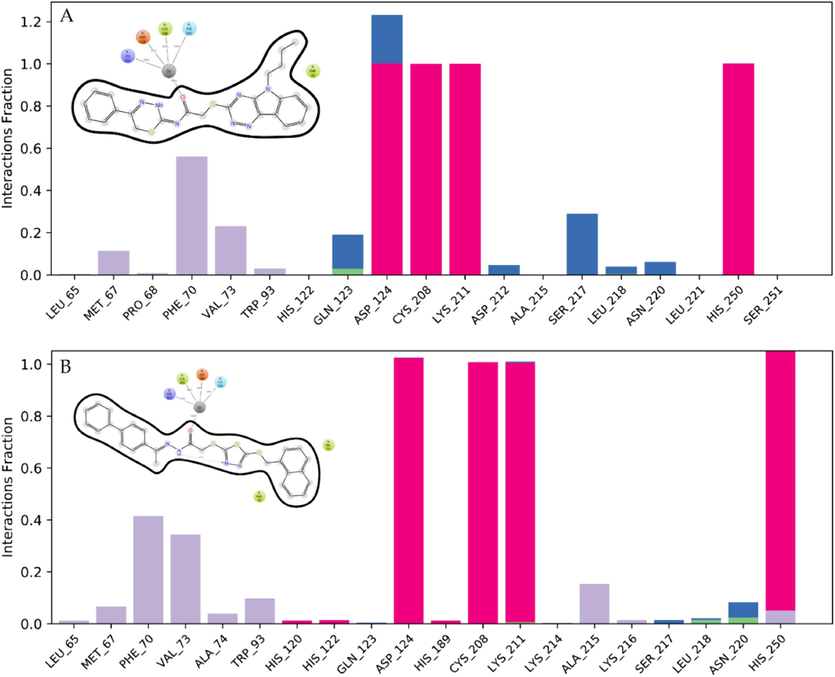
Ligand-protein interaction during MDS for STK115225 (A) and STK107343 (B). The interacting residues in NDM-1 are displayed as stacked bars.
3.5.1.3 Principal component analysis
PCA was employed to evaluate the dynamic behavior of the protein in both complexes (Fig. 8 A-B). This method aids in determining the collective motion of dynamics trajectories. Plotting the variance proportion against the Eigen values, which depict dynamic motions, was performed. Three principal components (PC) which cover the major fluctuations were plotted. The PCA analysis can demonstrate conformational changes in all groups through basic grouping in PC subspace (Khan et al., 2021). For the STK115225 complex, overall flexibility was recorded at 31.55 %. PC-1 elicited the highest variation of 15.35 %, while PC-2 and 3 had variabilities of 9.04 % and 7.16 %, respectively (Fig. 8A). With regards to the STK107343-complex, PC-1 also showed the maximum variation at 21.68 %. Corresponding variations of 6.8 %, and 5.71 % were observed for PC-2 and 3, respectively. Overall flexibility for the STK107343 complex was found to be 34.19 % (Fig. 8B).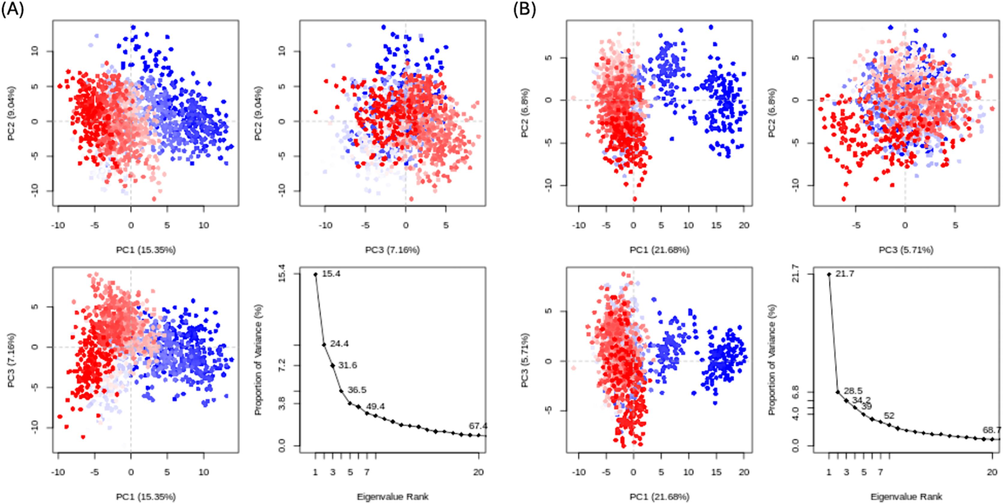
PCA of ligands STK115225 (A) and STK107343 (B) bound to NDM-1. Blue, white and red represents greatest, intermediate and least amount of flexibility movements. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.5.1.4 Cross correlation
Correlations amongst NDM-1 protein residues were assessed using a dynamic cross correlation matrix (DCCM; Fig. 9A-B). Most residues elicited positive correlations, but few and short-lived anti-correlated residues were also present. Overall, cross correlation matrix suggests that NDM-1 residues are highly correlated to in presence of the ligands during the MDS.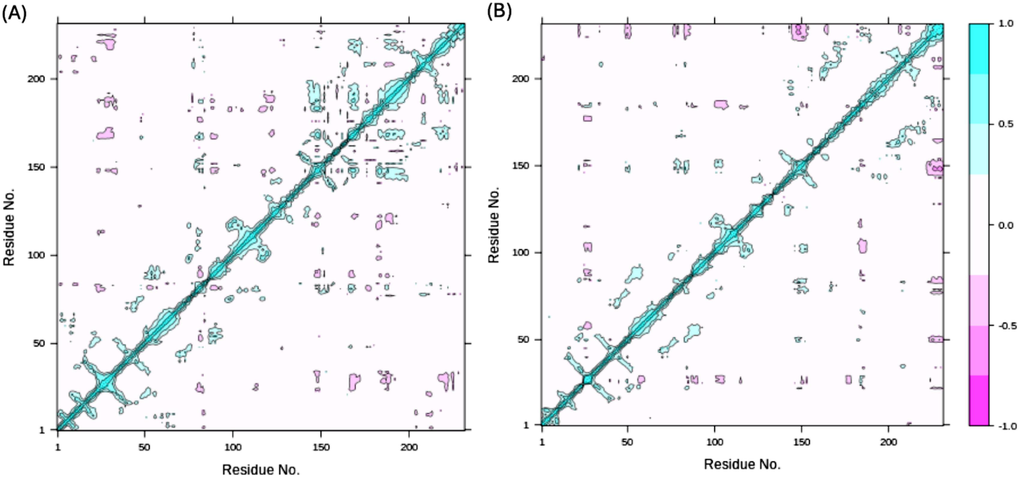
Dynamic cross correlation matrix (DCCM) of STK115225 (A) and STK107343 (B) complexed to NDM-1 protein. Cyan represents positively correlated residues, and magenta depicts the anti-correlated ones. Cyan diagonal lines in DCCM indicate positive correlations between topologically proximate residues. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.5.1.5 MM-GBSA calculations
MM-GBSA analyses were performed in order to calculate the total binding free energy (ΔGtotal) of the two ligands, STK115225 and STK107343 to NDM-1 protein. ΔGtotal is a good measure of the stability of protein–ligand complex (Du et al., 2011), and lower values indicate enhanced stability of the complex. ΔGtotal calculated by MM-GBSA model is dependent on the contributions of the various kinds of ligand–protein interaction energies, including van der Waals (ΔEvdW) and electrostatic (ΔEele) energies, and electrostatic contribution by generalized born (ΔGGB). ΔGtotal for the two complexes are depicted in Table 1.
The ΔEvdW contribution of STK107343 complex was more than STK115225 complex, and this was same for the electrostatic contribution. Similarly, generalized born contribution was higher for STK107343 when compared to the STK115225 complex. ΔGtotal was computed as −47.93 ± 0.31 kcal/mol for STK115225 complex, and −53.54 ± 0.43 kcal/mol for STK107343 complex. The contribution of each energy component in the calculation of ΔGtotal is depicted in Supplementary Fig. S1.
4 Discussion
Antibiotic resistance in pathogenic microbial has emerged as a significant global health issue. NDM-1 and its variants have recently attracted a lot of consideration because of their ability to hydrolyze practically all β-lactam antibiotics. These antibiotics include carbapenems, which have been traditionally thought to be last resort antibiotics. Horizontal gene transfer and enhanced spread of NDM-1 in newer bacterial species has aggravated the problems. In this regard, it is critical to identify and clinically evaluate NDM-1 inhibitors which may retard antibiotic resistance in pathogenic microbes and render current antibiotics usable and beneficial (Ahmed et al., 2021; Faheem et al., 2013; Rahman and Khan, 2020). In recent years, the utilization of investigational, current, and experimental drugs in drug development approaches has become increasingly important. Among them, high-throughput computational screening methods such as virtual screening of massive databases concentrating on different chemical principles, network pharmacology, and molecular docking have taken the lead (Löwer and Proschak, 2011; Thangavel and Albratty, 2022). Virtual screening of natural and synthetic molecules has emerged as a vital strategy for modern drug development (Walters et al., 1998). Virtual screening can be used for computational evaluation of novel small molecule chemicals from existent libraries for their binding effectiveness against target proteins. Chemical libraries are filtered using the docking approach, and compounds are ranked according to how well they bind (Lyne, 2002). In the present study, virtual screening in complementation with molecular docking has been employed in order to identify potent high-affinity binding ligands against NDM-1 protein.
The crystal structures of the NDM-1 protein were retrieved from PDB, and were aligned onto each other with reference to the binding pocket residues. The structures were aligned on each other to analyze the binding pocket residues. The RMSD difference was less than 0.5 which showed that there were not many deviations in the structures. Using the pharmacophore hypothesis, virtual screening of the small molecule ligand library obtained from the Vitas-M Laboratory database was carried out. A threshold phase screen score of 1.4 was utilized to identify possible hits, and 44 different compounds scoring higher than this threshold were chosen. These hits were analyzed for their binding to NDM-1 protein by molecular docking studies. This allowed us to predict the ligands’ affinity, orientation, and interaction when complexed with NDM-1. Docking results were evaluated as determined by glide gscores and molecular interaction parameters of the ligands (Silakari and Singh, 2021). In order to select high affinity binding hits and after setting a cutoff value of −7 kcal/mol, 4 hits (STK115225, STK107343, STK215880, and STK115575) were identified and further evaluated.
The docked poses of the ligands were superposed on the co-crystal and two compounds (STK115225, and STK107343) were selected for the protein–ligand stability analysis. STK115225 has been found to interact with the active site residues of NDM-1 such as His122, and His250. In addition, STK115225 was also involved in the interaction with second-shell residues of NDM-1 like Leu65, Val73, and Trp93. Similarly, STK107343 interacted with the catalytic residue Asp124 of NDM-1, along with other active site residues (His122, and His250), and second-shell residue (Trp93). Protein-ligand interactions usually involve receptor flexibility, which enables selectivity in ligand recognition. It follows that it is imperative to comprehend the conformational states of the ligand–protein complexes with the high affinity and specificity binding sites using techniques such as molecular dynamics simulations (MDS). MDS allows for the building of an ensemble of structures that may be used to compute thermodynamic potentials such as binding free energy with great precision (Salmas et al., 2015; Zimmermann et al., 2013). According to RMSF analyses, NDM-1 protein residues exhibited rigidity and did not exhibit substantial variations throughout the simulation duration, indicating high stability of the ligand–protein complex. Further, PCA revealed a large static number of hydrogen bonds. Additionally, the cross-correlation matrix was used to evaluate the correlation between the protein residues. Our results suggest that when the protein residues were bonded to their corresponding molecules during simulation, they showed a strong correlation with one another. Lastly, total binding free energies of the complexed ligands were calculated, and confirmed high stability of the complexes.
5 Conclusions
NDM-1 deactivates β-lactam antibiotics and leads to resistance against several antibiotics generally used in clinical settings. Hence, the present computational analyses explore promising small molecules with high binding affinities that could be exploited for designing NDM-1 inhibitors. Herein, Vitas-M laboratory database was screened for small molecules capable of binding to NDM-1 by generating a structure-based pharmacophore hypothesis, followed by molecular docking and dynamics simulations. The ligands, STK115225 and STK107343 were further analyzed for the stability and affinity of protein–ligand interactions. While we identified key compounds capable of targeting NDM-1, further experimental (in-vitro/-vivo) validation is required to confirm and extend our current findings (Saeidnia et al., 2016). However, in vitro and in vivo experiments are required to determine if these ligands (STK115225 and STK107343) can inhibit the biological function of the NDM1 protein and thus target antibiotic resistance in bacteria.
Ethical approval: This study is exempted from ethical approval as it is a type of in silico study and there is no involvement of patient/healthy subjects during the entire course of the study
Institutional Review Board Statement: Not applicable.
Informed Consent Statement: Not applicable.
CRediT authorship contribution statement
Shafiul Haque: Writing – review & editing, Writing – original draft, Visualization, Validation, Supervision, Software, Resources, Project administration, Methodology, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization. Darin Mansor Mathkor: Writing – review & editing, Writing – original draft, Visualization, Validation, Methodology, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization. Ayman K. Johargy: Writing – original draft, Visualization, Validation, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Hani Faidah: Writing – original draft, Visualization, Validation, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Ahmad O. Babalghith: Writing – original draft, Visualization, Validation, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Sumyya H. Hariri: Writing – original draft, Visualization, Validation, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Naif A. Jalal: Writing – original draft, Visualization, Validation, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Faraz Ahmad: Writing – review & editing, Writing – original draft, Visualization, Validation, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Farkad Bantun: Writing – original draft, Visualization, Validation, Methodology, Investigation, Formal analysis, Data curation, Conceptualization.
Acknowledgements
The authors extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project number ISP23-101. Few sentences of this manuscript were amended using ChatGPT, a language model developed by OpenAI, for text generation and linguistic analysis in this study. ChatGPT is a state-of-the-art natural language processing model based on the GPT-3.5 architecture (OpenAI, 2022).
Deputyship for Research & Innovation, Ministry of Education, Saudi Arabia - Project Number ISP23-101.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Identifying novel inhibitor of quorum sensing transcriptional regulator (SdiA) of Klebsiella pneumoniae through modelling, docking and molecular dynamics simulation. J. Biomol. Struct. Dyn.. 2020;1–11
- [CrossRef] [Google Scholar]
- Identifying novel inhibitor of quorum sensing transcriptional regulator (SdiA) of Klebsiella pneumoniae through modelling, docking and molecular dynamics simulation. J. Biomol. Struct. Dyn.. 2021;39:3594-3604.
- [CrossRef] [Google Scholar]
- Antiviral potential of some novel structural analogs of standard drugs repurposed for the treatment of COVID-19. J. Biomol. Struct. Dyn.. 2021;39:6676-6688.
- [CrossRef] [Google Scholar]
- Chemical profiling, antioxidant, enzyme inhibitory and molecular modelling studies on the leaves and stem bark extracts of three African medicinal plants. J. Pharm. Biomed. Anal.. 2019;174:19-33.
- [CrossRef] [Google Scholar]
- Bowers, K.J., Sacerdoti, F.D., Salmon, J.K., Shan, Y., Shaw, D.E., Chow, E., Xu, H., Dror, R.O., Eastwood, M.P., Gregersen, B.A., Klepeis, J.L., Kolossvary, I., Moraes, M.A., 2006. Molecular dynamics---Scalable algorithms for molecular dynamics simulations on commodity clusters, in: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing - SC ’06. ACM Press, New York, New York, USA, p. 84. doi: 10.1145/1188455.1188544.
- Structural basis of metallo-β-lactamase, serine-β-lactamase and penicillin-binding protein inhibition by cyclic boronates. Nat. Commun.. 2016;7:12406.
- [CrossRef] [Google Scholar]
- Updated functional classification of β-lactamases. Antimicrob. Agents Chemother.. 2010;54:969-976.
- [CrossRef] [Google Scholar]
- PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided. Mol. Des.. 2006;20:647-671.
- [CrossRef] [Google Scholar]
- Molecular modeling study of checkpoint kinase 1 inhibitors by multiple docking strategies and prime/MM–GBSA calculation. J. Comput. Chem.. 2011;32:2800-2809.
- [CrossRef] [Google Scholar]
- Biochemical Characterization of CTX-M-15 from Enterobacter cloacae and Designing a Novel Non-β-Lactam-β-Lactamase Inhibitor. PLoS One. 2013;8:e56926.
- [Google Scholar]
- Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem.. 2006;49:6177-6196.
- [CrossRef] [Google Scholar]
- The Bio3D packages for structural bioinformatics. Protein Sci.. 2021;30:20-30.
- [CrossRef] [Google Scholar]
- Revised Ambler classification of β-lactamases. J. Antimicrob. Chemother.. 2005;55:1050-1051.
- [CrossRef] [Google Scholar]
- Design and Prototyping of Genetically Encoded Arsenic Biosensors Based on Transcriptional Regulator AfArsR. Biomolecules. 2021;11:1276.
- [CrossRef] [Google Scholar]
- Approved drugs containing thiols as inhibitors of metallo-β-lactamases: Strategy to combat multidrug-resistant bacteria. J. Med. Chem.. 2015;58:3626-3630.
- [CrossRef] [Google Scholar]
- Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: a molecular, biological, and epidemiological study. Lancet Infect. Dis.. 2010;10:597-602.
- [CrossRef] [Google Scholar]
- Ten Years with New Delhi Metallo-β-lactamase-1 (NDM-1): From Structural Insights to Inhibitor Design. ACS Infect. Dis.. 2019;5:9-34.
- [CrossRef] [Google Scholar]
- Structure-Based Pharmacophores for Virtual Screening. Mol. Inform.. 2011;30:398-404.
- [CrossRef] [Google Scholar]
- Structure-based virtual screening: an overview. Drug Discov. Today. 2002;7:1047-1055.
- [CrossRef] [Google Scholar]
- Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des.. 2013;27:221-234.
- [CrossRef] [Google Scholar]
- Automatic Identification of Mobile and Rigid Substructures in Molecular Dynamics Simulations and Fractional Structural Fluctuation Analysis. PLoS One. 2015;10:e0119264.
- [Google Scholar]
- Discovery of Fungal Denitrification Inhibitors by Targeting Copper Nitrite Reductase from Fusarium oxysporum. J. Chem. Inf. Model.. 2017;57:203-213.
- [CrossRef] [Google Scholar]
- Phage Display Technique: A Novel Medicinal Approach to Overcome An tibiotic Resistance by Using Peptide-Based Inhibitors Against β-Lactamases. Curr. Drug Metab.. 2017;18:90-95.
- [CrossRef] [Google Scholar]
- A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys.. 2004;121:10096-10103.
- [CrossRef] [Google Scholar]
- In silico based unraveling of New Delhi metallo-β-lactamase (NDM-1) inhibitors from natural compounds: a molecular docking and molecular dynamics simulation study. J. Biomol. Struct. Dyn.. 2020;38:2093-2103.
- [CrossRef] [Google Scholar]
- From in vitro Experiments to in vivo and Clinical Studies; Pros and Cons. Curr. Drug Discov. Technol.. 2016;12:218-224.
- [CrossRef] [Google Scholar]
- Molecular Simulations of Solved Co-crystallized X-Ray Structures Identify Action Mechanisms of PDEδ Inhibitors. Biophys. J.. 2015;109:1163-1168.
- [CrossRef] [Google Scholar]
- How Molecular Size Impacts RMSD Applications in Molecular Dynamics Simulations. J. Chem. Theory Comput.. 2017;13:1518-1524.
- [CrossRef] [Google Scholar]
- Epik: a software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Aided. Mol. Des.. 2007;21:681-691.
- [CrossRef] [Google Scholar]
- Improving the Prediction of Absolute Solvation Free Energies Using the Next Generation OPLS Force Field. J. Chem. Theory Comput.. 2012;8:2553-2558.
- [CrossRef] [Google Scholar]
- Silakari, O., Singh, P.K., 2021. Chapter 6—Molecular docking analysis: Basic technique to predict drug-receptor interactions., in: Concepts and Experimental Protocols of Modelling and Informatics in Drug Design. pp. 131–155.
- Pharmacophore model-aided virtual screening combined with comparative molecular docking and molecular dynamics for identification of marine natural products as SARS-CoV-2 papain-like protease inhibitors. Arab. J. Chem.. 2022;15:104334
- [CrossRef] [Google Scholar]
- Recent research and development of NDM-1 inhibitors. Eur. J. Med. Chem.. 2021;223
- [CrossRef] [Google Scholar]
- Small molecule inhibition of the KRAS–PDEδ interaction impairs oncogenic KRAS signalling. Nature. 2013;497:638-642.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2024.103290.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




