

Translate this page into:
Molecular evolution of the pathogen recognition peptidoglycan proteins regulates the immune response against infectious diseases in Drosophila melanogaster
⁎Corresponding author. ishfaq.ahmad@iub.edu.pk (Hafiz Ishfaq Ahmad)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The study aimed to understand the molecular evolution of the pathogen recognition peptidoglycan (PGN) proteins and their role in regulating the immune response against infectious diseases in Drosophila melanogaster. D. melanogasterWe obtained the PGRP proteins from 11 different species of Drosophila and analyzed the different evolutionary trends that might be associated with them. We were able to identify the evidence of strong positive selection taking place for these proteins. We investigated the diversity and function of the PGN proteins in D. melanogaster and related species through a combination of bioinformatics approaches. They found that the PGN proteins have undergone rapid and diverse evolution, with some undergoing positive selection and others experiencing gene duplication and loss. The study also revealed that different PGN proteins play distinct roles in regulating the immune response to bacterial infections, with some responding specifically to certain types of bacteria. The research provides valuable insights into the evolution and function of the PGN proteins in the immune response of D. melanogaster. It highlights their potential relevance to pathogen recognition and immune defence in other organisms.
Keywords
Drosophila
PGRP
Adaptive immunity
Molecular evolution
Darwinian selection

1 Introduction
The innate immune system is the first line of defense against pathogens in all animals. It recognizes and responds to invading microbes through pattern recognition receptors (PRRs), specialized proteins that recognize conserved molecules in pathogens known as pathogen-associated molecular patterns (PAMPs) (Rasmussen et al., 2009). Peptidoglycan (PGN) is a major component of bacterial cell walls, and PGN recognition is a critical step in initiating the innate immune response. Several PGN recognition proteins have been identified in D. melanogaster, including PGRP-SA, PGRP-LC, and PGRP-LE (Wang et al., 2019). These proteins bind to PGN and trigger signaling pathways that activate the production of antimicrobial peptides and other immune effectors (Steiner 2004, Wang et al., 2019). Pathogen recognition peptidoglycan (PGN) proteins are critical in regulating the immune response against infectious diseases (Lu et al., 2020). These proteins recognize PGN, a major component of bacterial cell walls, through their pattern recognition receptors (PRRs). PGN recognition triggers signaling pathways that activate the production of antimicrobial peptides and other immune effectors, eliminating the invading pathogens. Different PGN recognition proteins respond to different types of bacteria, allowing for a diverse and specific immune response (Kurata, 2014).
Furthermore, the molecular evolution of PGN recognition proteins is complex, with some proteins undergoing positive selection and others experiencing gene duplication and loss (Demuth et al., 2006). Understanding the function and evolution of PGN recognition proteins is essential for developing new treatments for infectious diseases and gaining insights into animal pathogen recognition and immune defense mechanisms (Liang et al., 2022)..
Keeping immune systems effective in shifting pathogen repertoires and developing virulence mechanisms requires ongoing adaptation. Genes involved in the immune response can show a clear signal of adaptive evolution due to these dynamics (Liang et al., 2022). Due to the narrow scope of most studies, however, it has been tough to identify overarching trends. Drosophila responds to infections with both a cellular immune response and a cell-free (humoral) immunological response (Domingo, 2022). Differentiated populations of hemocytes engulf and melanize bigger parasites, including the eggs of parasitoid wasps, as part of the cellular immune response. The production of antimicrobial peptides (AMPs) and other effectors are triggered by the nuclear translocation of the NF-B transcription factors Relish, dorsal, and DIF in response to the identification of conserved microbe-specific compounds like peptidoglycan (Demir, 2022). Toll and imd pathways are crucially important, although other signaling cascades, such as JAK-STAT and JNK pathways, play supporting roles in this response (Tanji and Ip, 2005). In D. melanogaster, several PGN recognition proteins have been identified, including PGRP-SA, PGRP-LC, and PGRP-LE. These proteins play critical roles in regulating the immune response against different types of bacterial infections (Zaidman-Rémy et al., 2006). For instance, PGRP-SA is particularly effective against gram-positive bacteria, while PGRP-LC and PGRP-LE are more effective against gram-negative bacteria (Tanji et al., 2007). The different PGN recognition proteins activate distinct signaling pathways, leading to the production of specific immune effectors (Steiner, 2004). The molecular evolution of these proteins in D. melanogaster is complex, with some proteins undergoing positive selection and others experiencing gene duplication and loss (Robertson, 2019). Understanding the molecular evolution and function of PGN recognition proteins in D. melanogaster provides valuable insights into the mechanisms of pathogen recognition and immune defense in animals and can potentially lead to the development of new treatments for infectious diseases (Eleftherianos and Castillo, 2012). However, the genes involved in the immune response do not all evolve in the same way, and their divergent paths through time can help us infer how pathogens interact with various parts of the immune system (Sackton et al., 2007). All of the sequenced insect genomes include the PGRP and GNBP multigene families, suggesting that PRRs have higher evolutionary stability. In both the fruit fly family (Drosophila) (Hou et al., 2021) and the mosquito species (Aedes aegypti and Anopheles gambiae), there is scant evidence of genomic rearrangement. However, there has been a lot of change in the PGRP and GNBP gene families on the deeper lineages separating flies, mosquitoes, bees, and beetles (Papanicolaou et al., 2016). Evolutionary approaches are commonly used to understand the processes that drive protein evolution across different organisms. These approaches include phylogenetic analysis, positive selection tests, and comparative genomics (Clark et al., 2006).
Phylogenetic analysis is the process of determining the evolutionary relationships between species by comparing and contrasting their DNA (Xu et al., 2023). By comparing the genetic sequences of proteins from different species, researchers can infer the evolutionary history of these proteins and the selective pressures that have acted on them over time (Ahmad et al., 2022). Positive selection tests are commonly used to identify functionally important sites in proteins, such as those involved in ligand binding or protein–protein interactions (Ahmad et al., 2022). Comparative genomics involves the comparison of genome sequences from different species to identify similarities and differences in gene content and organization (Wei et al., 2002). These evolutionary approaches are particularly useful for studying the evolution of proteins involved in the immune system, such as the PGRP and MHC class I proteins. These proteins play critical roles in host defense and have undergone rapid evolution in response to changing pathogen pressures (Lazzaro, 2008). By using these evolutionary approaches, researchers can identify the key functional sites in these proteins and gain insights into the selective pressures that have driven their evolution over time. However, little is known about the molecular evolution of these proteins and their specific roles in regulating the immune response against different types of bacterial infections. In this context, understanding the molecular evolution and function of PGN recognition proteins is essential to gain insights into animal pathogen recognition and immune defense mechanisms. Therefore, this study aims to investigate the molecular evolution of PGN recognition proteins and their role in regulating the immune response against infectious diseases in D. melanogaster.
2 Materials and methods
The Kyoto Encyclopedia of Genes and Genomes (KEGG) database and the FlyBase annotations were searched for the amino acid and nucleotide sequences of D. melanogaster peptidoglycan recognition proteins (Wixon and Kell, 2000, Tweedie et al., 2009). The DNA sequences were retrieved by gene name or accession number, including FBgn0030310, FBgn0260458, FBgn0030695, FBgn0035975, FBgn0033327, FBgn0035976, FBgn0043576, FBgn0035977, FBgn0043577, FBgn0037906, FBgn0043578,FBgn0043575, and FBgn0035806. The aforementioned proteins encompass those that have been directly observed to fulfil an immunological function in D. melanogaster at the molecular level, as well as those that exhibit homology to established immune proteins found in D. melanogaster or other animal species. The MEGA 6 software was used to align their sequences using the ClustalW alignment tool (Tamura et al., 2013). The phylogenetic analysis was carried out with the help of MEGA 6, using the maximum likelihood approach. The bootstrap test uses a maximum likelihood method with 1000 value to assess branch length and topology for advanced log-likelihood values (Guindon and Gascuel, 2003).
2.1 Sequence analysis
Once the sequences are retrieved, they were aligned using software such as ClustalW, MAFFT, or MUSCLE. Multiple sequence alignment allows for the identification of conserved regions and amino acid changes between different species (Edgar, 2004). BioEdit was used to edit and arrange the sequence (Hall, 2004). A minimal number of functional loci was calculated by employing a conservational technique, and this estimate was based upon sequences that were shown to be translatable. All individuals from the sample species were assumed to be in the heterozygote state (Rosenberg et al., 2001).
2.2 Inference of recombination
The rate of recombination can be estimated using GARD software implemented in datamonkey, which uses coalescent theory to estimate the rate of recombination from sequence data (Kosakovsky Pond et al., 2006) Recombination analysis was performed using genetic recombination detection methods to calculate the genetic distance between sequences to detect recombination events. This method uses a sliding window approach to detect recombination events. In this method, the sequences are compared to a set of reference sequences, and recombination events are detected by the presence of incongruence in the phylogenetic trees (Kosakovsky Pond et al., 2006). The recombination events were detected by the presence of significant differences in the genetic distance between sequences. Maximum likelihood methods were based on the assumption that the sequences evolve under different evolutionary processes before and after the recombination event.
2.3 Tests for selection
Adaptive selection was identified in PGRP genes or genomic regions that have evolved under positive selection, meaning that they have undergone changes that improve their fitness and adaptation to their environment (Ahmad et al., 2020). PAML (Phylogenetic Analysis by Maximum Likelihood) software package was used to detect positive selection in protein-coding genes. PAML uses the codeml program to analyze the data (Yang, 2007). The codeml program compares the rates of synonymous and non-synonymous substitutions to detect positive selection. In this method the ratio of non-synonymous to synonymous substitutions (dN/dS) were compared in protein-coding genes. A dN/dS ratio greater than one indicates positive selection, while a ratio less than one indicates purifying selection (Bielawski et al., 2016). The output file generated by PAML were used to detect positive selection by calculating likelihood log ratios (2ΔlnL) using χ2 distribution (Moretti et al., 2012).. Datamonkey is a webserver was further used for detecting positive selection in protein-coding sequences (Pond and Frost, 2005). Different methods including FEL (Fixed Effects Likelihood), REL (Random Effects Likelihood), FUBAR (Fast Unconstrained Bayesian AppRoximation), and MEME (Mixed Effects Model of Evolution) available on Datamonkey were used to detect positive selection (Weaver et al., 2018). FEL, REL, and MEME are all likelihood-based methods that use a phylogenetic tree to detect positive selection. FEL and REL identified the constant selective pressures acting on the sequence across the entire tree or at each site, respectively (Delport et al., 2009). MEME, on the other hand, allowed for different selective pressures to act on different sites in the sequence (Murrell et al., 2012). FUBAR, on the other hand, is a Bayesian method that used a Markov Chain Monte Carlo (MCMC) algorithm to estimate the posterior probability of positive selection at each site in the sequence (Murrell et al., 2013). The Selecton server was used to further confirm positive selection in protein-coding sequences data. The MCMC model implemented in Selecton Server used a Bayesian approach to estimate the posterior probabilities of different selection pattern at each site in the protein-coding sequences. It allows for both positive and negative selection, as well as the possibility of no selection, at each site. The MCMC algorithm samples from the posterior distribution to estimate the probability of each selection region (Stern et al., 2007).
2.4 Phylogenetic analysis
MEGA (Molecular Evolutionary Genetics Analysis) version 10.0.5 was used to create phylogenetic trees using the maximum likelihood method. The initial tree construction was performed using the neighbor-joining method, and the tree topology was evaluated through the application of the maximum likelihood method. One thousand bootstrap replications were conducted to evaluate the resilience of the tree topology. The species tree produced by TreeBeST serves as a point of reference for evaluating gene trees and other phylogenetic trees. The authors of the study conducted phylogenetic network analysis to detect instances of reticulation in the proteins' evolutionary lineage (Jin et al., 2006).
The second stage of determining positive selection is to identify which amino acid sites in a protein sequence have undergone positive selection. Positive selection refers to the process by which mutations that confer a fitness advantage become more frequent in a population over time (Smukowski Heil, 2023). Positive selection has been linked to the evolution of proteins by altering particular amino acid residues in ways that have an effect on the protein's function or structure. The rate of synonymous (i.e., silent) versus non-synonymous (i.e., amino acid altering) substitutions at each amino acid position was compared as a proxy for the presence of positive selection. Positive selection may be present if the rate of non-synonymous substitutions at a given site is much higher than the rate of synonymous substitutions at that site (Mayrose et al., 2007). Moreover, comparing the amino acid sequences of homologous proteins from different species, looked for evidence of convergent evolution at specific sites. The two distantly related species have evolved similar amino acid changes at the same site, this can suggest that positive selection has occurred (Russell, 1998).
2.5 Conservation analyses
The conservation analysis of pathogen recognition peptidoglycan proteins (PRRs) was performed using the Consurf server (Ashkenazy et al., 2016). The amino acid sequences of the PRRs were obtained from public databases such as GenBank or UniProt used as input for the analysis. The Consurf server is a bioinformatics tool used to perform conservation analyses of protein sequences. It is commonly used to identify conserved regions in protein structures, which can help to identify functional domains and residues that are important for protein function (Goldenberg et al., 2009). The output of the Consurf server is a conservation score for each residue in the protein sequence, ranging from 1 (most variable) to 9 (most conserved). Conserved residues are often represented as a different color or shape in a protein structure visualization, which can help to identify functionally important regions.We investigated the degree to which the genomic areas close to the PGLYRP gene were conserved in synteny among Drosophila species. The PGLYRP protein from drosophila was analyzed for evolutionary conservation in amino acid residues using the ConSurf library (consurf.tau.ac.il/) (Celniker et al., 2013). These amino acids have a higher degree of conservation and are found in more enzymatic nooks or protein–protein interactions than others. Because they disrupt both the function and the structure of the protein, changes in conserved amino acids are an even more dangerous form of polymorphism within a protein than polymorphisms in more flexible regions (Glaser et al., 2003).
2.6 3D protein modeling and structural analysis
The crystal structure of the human compound PGRP domain (PDB assembly number 4D8A) is a homologous structure to the PGRP protein of D. melanogaster (Burley et al., 2017). Therefore, it was used as a template to model the 3D structure of the Drosophila PGRP protein. Protein modeling was performed using software such as SWISS-MODEL (Waterhouse et al., 2018) and I-TESSAR (Zheng et al., 2021), and Phyre2 (https://www.sbg.bio.ic.ac.uk/phyre2/html) which utilize comparative modeling techniques to generate a 3D structure based on the template structure. The sequence of the Drosophila PGRP protein was used as input into the software, along with the template structure, and the software generated a model of the Drosophila protein based on the alignment between the two sequences. Once a 3D model of the Drosophila PGRP protein has been generated, it was subjected to further refinement and validation using software such as PyMOL (DeLano, 2002). These programs allow for the visualization and manipulation of the protein structure, as well as the analysis of various structural features such as hydrogen bonding, salt bridges, and solvent accessibility. To analyze the residue-residue interactions in protein structures, including receptor-ligand interactions and hydrogen-bonding epitopes we used protein-protien interaction network approaches (Zhang et al., 2010). LigPlot tool was used for analyzing the protein–ligand interactions, including protein–protein interactions. It generated schematic diagrams of protein–ligand interactions, highlighting the key residues involved in the interaction (Laskowski and Swindells, 2011). LigPlot generated schematic diagrams of protein–ligand interactions, highlighting the key residues involved in the interaction. The diagrams provided a visual representation of the interactions between the proteins and the types of interactions that occur, such as hydrogen bonds, hydrophobic interactions, or electrostatic interactions (Norris et al., 2012).
3 Results
The study on the molecular evolution of pathogen recognition peptidoglycan proteins (PRRs) in D. melanogaster aimed to understand the evolutionary mechanisms that regulate the immune response to infectious diseases. The study analyzed the genetic sequences of PRRs in several Drosophila species and examined how changes in PRR genes affect the immune response of Drosophila to bacterial infections. The study found that PRR genes are highly conserved across Drosophila species, suggesting that they have important functions in host-pathogen interactions. Ths study also identified specific regions of the PRR genes that were under positive selection, indicating that they are involved in the evolution of the immune response to bacterial infections. The study further demonstrated that changes in PRR genes can affect the immune response of Drosophila to bacterial infections. Specifically, the researchers showed that mutations in the PGRP-SA gene, which encodes a peptidoglycan recognition protein, can affect the sensitivity of Drosophila to bacterial infections. Overall, the study highlights the importance of PRRs in regulating the immune response to bacterial infections in D. melanogaster and provides insights into the evolutionary mechanisms that shape the immune response to infectious diseases. The findings may have implications for the development of novel strategies for the treatment and prevention of infectious diseases in both animals and humans.
3.1 Adaptive evolution of peptidoglycan recognition proteins
The adaptive evolution of PGRPs revealed to understand how these proteins have evolved to recognize and respond to a diverse array of bacterial pathogens. Our study has identified several regions of PGRP genes that are under positive selection, indicating that they have evolved rapidly in response to selective pressures imposed by bacterial pathogens. These regions are often located in the peptidoglycan-binding domain of PGRPs, suggesting that they play a key role in the recognition of bacterial cell wall components. We utilized a variety of site models to determine which genes were being positively selected across vertebrate species. We used the phylogenetic tree in the dataset to evaluate many gene model comparisons. Codons in genes undergoing positive selection were categorized using a probabilistic technique that compared models based on different ratios. To conduct the positive selection test, two sets of models (M1a; M2a and M7; M8) were employed. The likelihood test value for M1a vs. M2a was 0 (p > 0.05). However, the likelihood ratio test results for PGLYRP were as follows: 2lnL = 64.28, 2lnL = 4.9283, 2lnL = 49.31, and 2lnL = 68.65 (Table 1). Multiple correlating approaches from the HyPhy package were then used to assess the evidence for positive selection (Table 1). Through MEME, FEL, and SLAC analyses, we inferred evolutionary signs consistent with positive selection based on global relationship values. As a result, sites discovered by multiple methods (i.e., those that were consistent with two or more methods) were prioritized for positive selection. Our results provided compelling proof that these genes have been subjected to successful genetic selection in Drosophila species. Bayesian analysis was utilised to classify sites experiencing selective pressure by estimating posterior probabilities for each codon. Positive selection is more likely to occur in higher-probability sites, as seen in Fig. 1, when either > 1 or. With the help of BEB analysis, we were able to pinpoint several positively selected locations within these proteins, with most of them having retrospective probabilities above 95%. Further validating the good results of selection, we used the Selecton server and the Mechanical-Empirical Hybrid model to confirm positive selection on individual sites. During the course of the organism's evolution in response to selective pressure, we found a large number of previously unknown sites (Fig. 1).
Residue
p-value
Bias term
Proportion of affected sites
Directionally evolving sites
F
0.0006
3.4
2.30%
4
L
0.0003
2.08
3.98%
4
M
0
10,000
0.19%
1

Domain structure, alignment, and positively selected sites in the 3D structure of PGRP of Drosophila. (A) Domain structure of PGRP of D. melanogaster, consisting of a transmembrane domain and a PGRP domain. (B) Alignment of PGRP sequences from multiple Drosophila species, showing conservation of key amino acid residues involved in peptidoglycan binding (red), and positively selected sites (yellow) identified by evolutionary analyses. (C) Crystal structure of PGRP of D. melanogaster, showing the location of the positively selected sites and the conservation score. This figure provides a comprehensive overview of the domain structure, sequence conservation, and 3D structure of the PGRP protein in Drosophila, highlighting the key features involved in its function and evolution. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
In PGLYRP, positively selected sites were discovered with an average frequency of 8. In total, we concluded that each gene possesses two positively selected sites within the peptidoglycan-binding type II amidase (PGRP) domain. This domain is structurally comparable to the type II amidase found in bacteria. Based on the crystal structure of the human compound PGRP domain, positions 325, 326, 327, 340, 345, 351, 352, 353, 359, and 372 on the dimerization surface are important for ligand binding. Compared to neutral evolution models (M7), the M8 model that accommodates positive selection performs better in likelihood calculations. There was an acknowledgment of the positive selection of sites, and these sites are listed in (Table 2). In total, we identified 12 positively selected codons throughout the sampled species, 3 of which are homologous to positively selected codons in other bird species, and 1 of which is a perfect match to a human peptide binding area (Table 2). Conservation analyses of PRRs using the Consurf server can provide valuable insights into the evolution and function of these proteins in host-pathogen interactions. By identifying conserved regions and residues, researchers can gain a better understanding of the mechanisms of PRR recognition and the development of immune responses to bacterial infections. Conservation analyses can also be used to identify the selective pressures acting on PRR genes. For example, a study of the TLR4 gene in primates found evidence of positive selection, suggesting that this gene has been subject to strong selective pressures in response to pathogen pressure. In addition to changes in the peptidoglycan-binding domain, studies have also identified adaptive changes in other regions of PGRP genes that affect the activity and specificity of the proteins.
Site
Composition
MRCA Residue
Inferred Substitutions
DEPS EBF
Selection kind
1
M26
L
L0↔2M
M:>105
M:(Partial) selective sweeps
6
L7E5A4I2S2N2K1F1V1D1
E
A1↔0E
L: 107.1
L:Convergent evolution/Repeated Substitutions
A3↔0M
D1↔0E
D0↔1S
E0↔2I
E0↔1K
E0↔4L
E3↔3M
E0↔1N
F1↔0M
L3↔0M
M0↔1N
M0↔1S
M0↔1V
46
L8A5I2E2M2S2G1Y1W1V1R1
A
A0↔2E
L: 414.9
L:Convergent evolution/Repeated Substitutions
A0↔1G
A0↔2I
A0↔7L
A0↔2M
A0↔1R
A0↔2S
A0↔1V
A0↔1W
A0↔1Y
120
F6D5K4P3S2A1I1G1E1L1W1
D
A1↔0D
F:7148.1
F:Convergent evolution/Repeated Substitutions
A0↔1P
D0↔1E
D0↔3F
D0↔1G
D0↔1I
D0↔3K
D2↔2M
D0↔2P
D0↔1S
D0↔1W
F3↔0M
K1↔0M
L1↔0M
M0↔1S
150
L9D5A3Q2G2K1I1Y1P1
D
A1↔0D
L:2138.3
L:Convergent evolution/Repeated Substitutions
A2↔0M
D0↔1G
D0↔1I
D0↔1K
D0↔7L
D2↔3M
D0↔1P
D0↔1Q
G1↔0M
L2↔0M
M0↔1Q
M0↔1Y
165
G7F4A2I2R2T2K1D1E1L1V1Q1
G
A1↔0G
F: 126.4
F:Convergent evolution/Repeated Substitutions
A1↔0M
D1↔0G
E1↔0G
F3↔0G
F1↔0M
G0↔2I
G0↔1K
G0↔1L
G4↔3M
G0↔2R
G0↔1T
M0↔1Q
M0↔1T
M0↔1V
230
I6F4P2K1N1D1S1
I
D1↔0I
F: 661.2
F:Convergent evolution/Repeated Substitutions
F3↔0I
F1↔0M
I0↔1K
I1↔0L
I4↔3M
L0↔1M
M0↔1N
M0↔2P
M0↔1S
284
L10C1Y1P1
L
C1↔0L
L: 183.5
L:Frequency dependent selection
L1↔1M
L0↔1P
L0↔1Y
301
T5F3L2G1E1
T
E1↔0T
F: 185.3
F:Convergent evolution/Repeated Substitutions
F2↔0M
F1↔0T
G0↔1M
G1↔1T
L1↔0M
L1↔0T
M3↔4T
3.2 Selection analysis
To determine whether adaptive selection is occurring in the PGRP proteins, we used various software such as PAML (Phylogenetic Analysis by Maximum Likelihood) to perform likelihood ratio tests (LRTs). These tests compared the fit of different models of sequence evolution, including models that allow for positive selection, to determine whether positive selection is occurring at particular sites in the protein sequence. The identification of sites under positive selection provide insights into the functional evolution of the PGRP protein and its role in adaptation to changing environmental conditions or in host-pathogen interactions in Drosophila species. Furthermore, Using an EBF threshold of 100, we found that only a small fraction of sites (3%) were detected as evolving in a particular direction for each genomic region. A fixed effects likelihood (FEL) analysis yielded similar percentages for the number of locations suspected of being subject to diversifying positive selection. Most existing approaches for detecting positive selection, including FEL, are based on the equal rates model for replacing amino acid residues and a codon substitution model. Therefore, positive selection can be identified if the rate of nonsynonymous substitution at a site is higher than the rate of synonymous substitution at the same site on average (considering all possible residue pairs). FEL makes direct estimates of both rates at each location and uses an LRT to determine whether they are comparable. When looking at the results site-by-site (Tables 2 and 3), one of the most important things to understand is that sites subjected to directionally selected selection are overwhelmingly different from sites found to be subjected to diversifying selection, with a few exceptions in PGRP. In addition, there are a few sites that, based on a conventional dN/dS analysis, appear to be undergoing purifying selection but are nevertheless evolving in a particular direction. We also identified Directional selection in PGRP protein of Drosophila species. Directional selection is a type of natural selection that occurs when one allele is consistently favored over another, leading to a shift in the frequency of that allele over time. Directional selection can lead to the rapid evolution of specific traits, including those involved in the immune response. We have identified sites that are under directional selection in the PGRP genes of D. melanogaster. These sites are located in the peptidoglycan-binding domain of the protein and are thought to be important for the recognition of specific bacterial pathogens. Convergent evolution is the independent evolution of similar traits in different lineages. In the case of the PGRP of D. melanogaster, there is evidence of convergent evolution in the repeated substitutions of amino acids in the protein. Our study have shown that the PGRP of D. melanogaster has undergone repeated substitutions at specific amino acid sites. These substitutions are thought to be driven by positive selection, which means that they provide some sort of advantage to the organism. The repeated substitutions have occurred independently in different lineages of D. melanogaster, suggesting convergent evolution (Table 2).
Model
Parameter estimates
PAML
IFEL
REL
MEME
FUBAR
M1 Nearly Neutral (2 categories)
P1 = 0.52191 P2 = 0.47809
0
289,326, 335, 340, 352, 359, 372, 466
366, 345,359, 426, 441, 466
326, 345, 359, 372, 379, 426, 466, 492
326, 335, 340, 345, 359, 368, 372, 411, 426, 466, 479, 492
ω1 = 0.16490 ω2 = 1.00000
M2 Positive Selection (3 categories)
P1 = 0.52191 P2 = 0.34105 P3 = 0.13704
325, 326, 327, 335, 340, 351, 352, 353
ω1 = 0.16489 ω2 = 1.00000 ω3 = 1.00000
M3 discrete
(3 categories)P1 = 0.23174 P2 = 0.65157 P3 = 0.11670
325, 326, 327, 349, 351, 352, 353, 366, 386, 426
ω1 = 0.07398 ω2 = 0.42630 ω3 = 3.63400
M7 beta
(10 categories)p = 0.90528 q = 1.10286
0
M8 beta & w > 1 (11 categories)
P0 = 0.90380 p = 1.17592 q = 1.93637
P1 = 0.09620 ω1 = 4.44414
325, 326, 327, 340, 345, 351, 352, 353, 359, 372
3.3 Codon model selection
The molecular processes of phenotypic adaptation were investigated by examining peptidoglycan recognition proteins of Drosophila species. We explored how these proteins have evolved to select particular amino acid locations by using a variety of codon models available on the DATAMONKEY online service. Our findings revealed that the amino acid replacement rates varied at different points in the amino acid sequences of these proteins, indicating adaptive evolution. Specifically, we discovered that these proteins had undergone adaptive evolution with varying replacement rates at different locations in their amino acid sequences. For PGLYRP, based on various ratio groupings, the average substitution rates were roughly 1.38, 0.54, and 0.12, respectively, with the lowest being 0.12 for several amino acid sites in PGLYRP (as shown in Fig. 3). This analysis utilized a subset of the codon model that relied on a modified Bayesian Information Criterion and the probability log (mBIC).Using the physicochemical factors linked with combined empirical codon and transition/transversion (Fig. 3). Using the mBIC test and GAs, we were able to implement a standardized multi-rate test on a data set successfully. It is involved in the recognition of bacterial peptidoglycan and the triggering of immune responses. We have shown that the amino acid sequences of the PGRP domain have undergone positive selection, which is a process in which advantageous mutations are fixed in a population due to their ability to confer some selective advantage. Specifically, certain amino acid residues in the PGRP domain have experienced a higher rate of substitution than would be expected by chance, suggesting that they have been under positive selection. The adaptive evolution of the PGRP domain is thought to be driven by the co-evolutionary arms race between insects and bacteria. This leads to the evolution of new amino acid sequences in the PGRP domain that are better able to recognize and respond to bacterial peptidoglycan.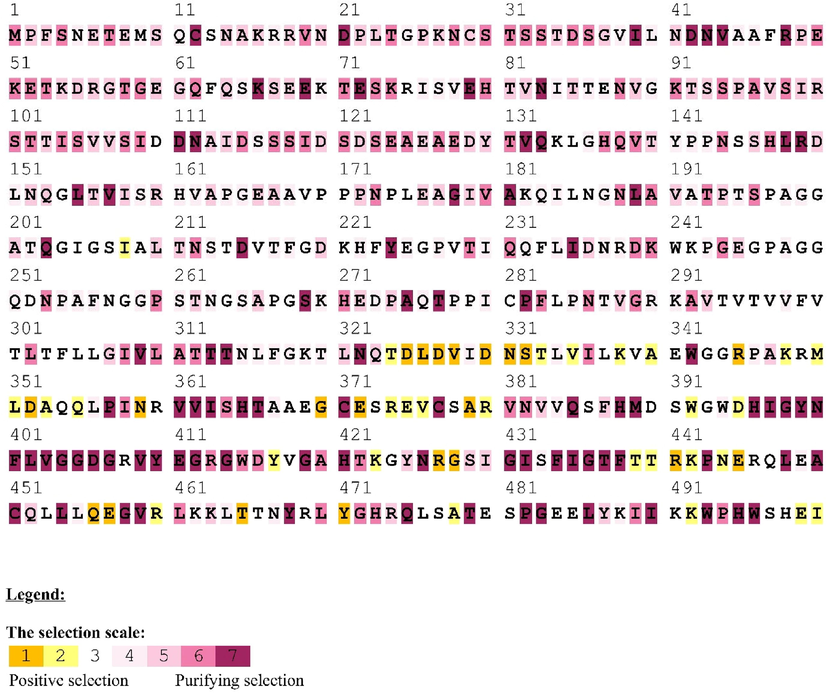
Positively selected amino acid sites in the PGRP protein sequence of Drosophila species displayed in the outcomes of the Selecton server analysis. These locations were marked in some way to draw attention to them. Statistical metrics (1–9) are also provided, such as the level of support for positive selection at each location.
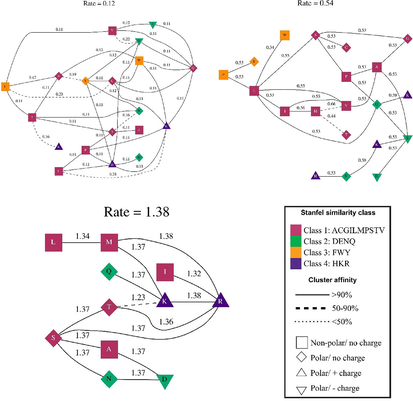
Shows the substitution rates of amino acid sequences of the peptidoglycan recognition protein (PGRP) domain that had undergone adaptive evolution. The analysis was based on various ratio groupings using a subset of the codon model that relied on a modified Bayesian Information Criterion and the probability log (mBIC). The average substitution rates for PGLYRP were approximately 1.38, 0.54, and 0.12, respectively, with the lowest rate being 0.12 for certain amino acid sites in PGLYRP. These results indicate that different locations in the amino acid sequences of these proteins have undergone varying replacement rates, suggesting adaptive evolution.
3.4 Functional analysis of PGRP protein
Protein-protein interactions (PPIs) are crucial for the function of PGRP proteins in the immune response. PGRPs can interact with other immune-related proteins, such as Toll-like receptors (TLRs) and Nod-like receptors (NLRs), to activate downstream signaling pathways that lead to the production of cytokines and other immune response mediators. Different types of proteins and their secondary structures have different patterns of inter-residue interactions. In this study, we will describe these interactions' role in proteins that belong to various structural classes and folds, including globular, membrane, and other protein folds. The -carbon atom of a protein residue serves as a symbol for the entire protein molecule. Distances are calculated between the -carbon atom of the first (N-terminal) residue and all other carbon atoms in the protein molecule. We did this using Ligplot graphs based on the different stances (Figs. 4 and 5). These diagrams illustrate the interactions between the drug (shown here as a ball and stick) and the residues located in the pockets that are close by. The presence of hydrogen bonds is denoted by a dashed line that connects the atom of the drug ligand to the atom of the residue that lines the pocket. This line is also represented as a ball and stick in the instances in question. Van der Waals contacts can be seen as “fans” of lines circling each residue and pointing in the direction of the drug. The drug itself has fans pointing in the opposite direction, towards the residues lining the pocket with which it interacts. This investigation uncovered a pocket of particular importance because it is situated at an interface between two chains, A and X, of the protein (Fig. 4). Comparable residues in A and B or A and C do not include this pocket, which means it is distinct from those locations. This is because chains A and X both have their mobile domains in the up position, in contrast to chain A, which has its mobile domain in the down position. The pocket can be found between two domains, specifically in the protein's chain A flanking area. The PGRP (peptidoglycan recognition protein) receptor binds to bacterial peptidoglycan and triggers an immune response in insects, including D. melanogaster. The binding pocket of the PGRP receptor is the region of the protein that directly interacts with the peptidoglycan. Interactions between protein residues in the binding pocket of the PGRP receptor are critical for the specificity and affinity of the protein-peptidoglycan interaction. The binding pocket contains a number of amino acid residues that interact with the peptidoglycan, and these residues are highly conserved across different species of insects. We have identified several key residues in the binding pocket of the PGRP receptor that are important for the interaction with peptidoglycan. For example, in D. melanogaster, the residues Y63, D132, and R134 have been shown to be critical for the binding of peptidoglycan (Fig. 4 and Fig. 5). These residues form hydrogen bonds and other interactions with the peptidoglycan, stabilizing the complex and allowing for efficient recognition. In addition to these key residues, there are also interactions between other residues in the binding pocket that contribute to the overall stability and specificity of the protein-peptidoglycan complex. For example, there may be interactions between hydrophobic residues that help to anchor the peptidoglycan in the binding pocket, or between charged residues that contribute to electrostatic interactions.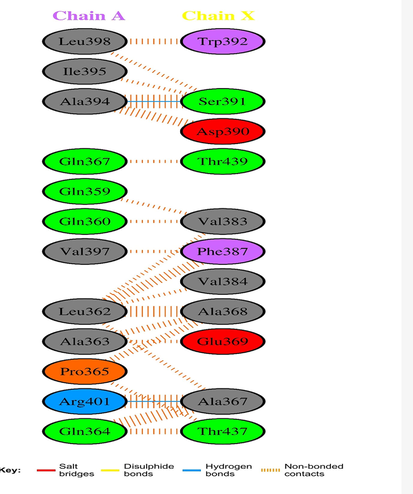
The receptor's and the hydrogen-bonding epitopes' residue-residue interactions are denoted by green and blue, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
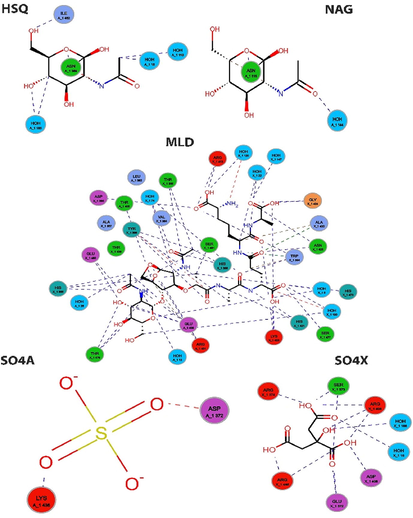
Interactions between protein residues in the binding pockets. The PGRP receptor's residues interact with ligands, and the hydrogen-bonding-related epitopes are denoted by the colors with ligands. The hydrogen-bonding-related epitopes are denoted by blue and green, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
4 Discussion
The pathogen recognition peptidoglycan proteins (PGRPs) are critical components of the innate immune response in D. melanogaster, playing an essential role in recognizing and responding to infectious pathogens (Leclerc and Reichhart, 2004). Our analysis of the molecular evolution of PGRPs in D. melanogaster provides insights into the adaptive mechanisms that have enabled this species to respond to a diverse range of pathogens. We found that the PGRP gene family is ancient and diverse, with members separated by long evolutionary histories. The chromosomal localization, DNA sequence divergence, and variable intron positions of these genes further indicate their long evolutionary history (Roesner et al., 2005). However, despite this diversity, it was observed that the PGRPs are widespread across the genome of D. melanogaster, occupying eight chromosomal positions ranging from the first to the third chromosome (Schmid, 2014). Our findings suggest that the PGRP genes have undergone adaptive evolution, with different locations in the amino acid sequences of these proteins exhibiting varying replacement rates. This suggests that the evolution of PGRPs is driven by the need to recognize and respond to a diverse range of pathogens. Furthermore, we observed that certain PGRP genes, such as the PGRP-SC genes, exhibit high similarity in both DNA sequence and tissue expression patterns, potentially arising from recent duplications. These results suggest that the molecular evolution of PGRPs is shaped by both long-term evolutionary processes and more recent adaptive mechanisms. The diversity and widespread distribution of PGRPs across the genome of D. melanogaster, combined with the varying rates of replacement in different locations of the amino acid sequences of these proteins, highlight the importance of this family of genes in regulating the immune response against infectious diseases (Gerland et al., 2017). Our findings provide a foundation for future studies exploring the molecular mechanisms underlying the evolution and function of PGRPs in D. melanogaster and other organisms.
The adaptive evolution of PGRPs reflects the ongoing arms race between host organisms and bacterial pathogens. As bacteria evolve to evade host immune responses, host organisms must adapt their immune systems to recognize and respond to new and emerging pathogens. Understanding the adaptive evolution of PGRPs and other innate immune proteins may have important implications for the development of novel strategies for the treatment and prevention of infectious diseases (Obbard et al., 2009). Alleles with a beneficial mutation that maintain an individual's fitness are preserved by positive selection. Our study's classical selection tests (Tajima's D, Fu & Li's D*, and Fu & Li's F*) indicated no significant departure from neutral selection or balance selection. When dealing with such a large range of possible outcomes, the effectiveness of tried-and-true approaches for identifying selection is severely diminished (Montaño et al., 2011). Variation in amino acid sequence can alter protein function as positively selected sites tend to accumulate more non-synonymous than synonymous changes (Li et al., 2009). Our research revealed that the peptide binding region (PBR) and non-PBR sections of the PGRP gene exhibited different selection patterns in functional sequences. In the PBR of Turdus atrogularis, non-synonymous substitution was more common than synonymous substitution (dN/dS = 1.99), and this trend was consistent across all tested species (dN/dS = 0.884), indicating that intracellular pathogens exert stronger selection pressure than extracellular pathogens. House sparrows and golden pheasants showed positive selection at their MHC PBR (PBR dN/dS = 1.55 and 1.45, respectively), while non-PBR regions exhibited lower dN/dS values (0.51 and 0.91, respectively). Nine, 29, 64, and 88 of the 12 codons identified as positively selected across all species examined using likelihood techniques employing PAML had homologs that were positively selected in other passerine species. It is important to remember that when alleles are combined across loci, selection detection tests are drastically lowered, leading to results that may be conservative but are less likely to be false positives (Fijarczyk and Babik, 2015). Innate defensive mechanisms are crucial for protecting organisms from invading pathogens, and PGRP signaling is an efficient pathogen identification system that is engaged in this process. Several patterns in PGRP proteins have been preserved from one Drosophila species to another, suggesting their importance in pathogen detection (Shokal and Eleftherianos, 2017).Positive selection is an essential factor in gene evolution, which contributes to the rapid adaptive development of genes. Positive selection has been observed at varying levels in newly generated genes, and it is widely held that this process causes the emergence of novel genetic material (Zhang, 2003). Studies of PGRPs genes for selection show that this gene has been under positive selection pressure. This suggests that PGRPs genes, which may have arisen due to gene duplication, are a novel gene that seeks to adjust to a novel environment. As a result, the gene's coding sequence has undergone ongoing alteration due to positive selection and the accumulation of beneficial mutations (Taylor and Raes, 2004). This occurs when a group of animals experiences a shift in environmental conditions or pressure, forcing them to adapt by creating a new gene with characteristics tailored to their new surroundings (Sackton et al., 2007). For example, a study by Lazzaro et al. (2002) identified several sites in the PGRP-SA gene that are evolving rapidly and are likely to be under directional selection (Lazzaro, 2002). Another study by Garsin et al. (2001) identified several sites in the PGRP-LC gene that are under directional selection (Garsin et al., 2001). These sites are also located in the peptidoglycan-binding domain of the protein and are thought to be involved in the recognition of bacterial peptidoglycan. More recent studies using advanced computational methods, such as phylogenetic analysis and codon-based models, have identified additional sites under directional selection in PGRP genes of D. melanogaster. Overall, the identification of sites under directional selection in PGRP genes of D. melanogaster provides insights into the molecular evolution of the innate immune system and the mechanisms by which host organisms adapt to new and emerging bacterial pathogens.
The empirical Bayes method was used to determine the likelihood ratio test (LRT) at each branch site and identify the diversifying selection sites. The empirical Bayes method-based mixed-effects model evolution (MEME) was used to characterize the diversifying selection of the PGLYRP gene (Fig. 1). MEME was used to examine adaptive evolution at the genetic level, providing information about the dispersal from one location to another and from one branch to another. The MEME detected different codon locations with a p-value of 0.01 when subjected to episodic diversification selection (Table 1). The model was used to determine the ratio of synonymous to non-synonymous substitutions and how important coding sites with values ω > 1 were in identifying the sites under selection diversification. Using MEME, we calculated a maximum probability estimate for each codon in PGRP gene of each Drosophila species (Table 1). Since purifying or natural selection was so common, it wasn't easy to detect the natural selection of episodes with a fleeting cycle of adaptive evolution. However, positive selection with responsive checks does reveal this (Barash, 2007). To assess the degree of uncertainty in the posterior gene and site-specific distribution, the Markov Chain Monte Carlo (MCMC) algorithm was combined with the fast unconstrained Bayesian approximation (FUBAR). The PGLYRP gene was subjected to constant diversifying selection using the FUBAR method, yielding four sites in PGLYRP (Table 1). We observed that the most favorably chosen locations were preserved across mammalian clades during evolution (Fig. 2). NNA (neural network algorithm) residues have been shown to expose or hide a large number of retained amino acids, presenting a positive range of signals. The repeated substitutions in the PGRP of D. melanogaster are believed to be adaptive changes that allow the protein to recognize and bind to a wider range of bacterial peptidoglycans. This is important because different types of bacteria have different peptidoglycans, and the ability to recognize a wider range of peptidoglycans may provide a selective advantage in a changing environment (Fijarczyk and Babik, 2015). Overall, the repeated substitutions in the PGRP of D. melanogaster are an example of convergent evolution driven by positive selection. The changes in the protein provide an adaptive advantage by allowing the recognition of a wider range of bacterial peptidoglycans, and these changes have occurred independently in different lineages of D. melanogaster.
4.1 Evolutionary fingerprinting of PGRP genes
Evolutionary fingerprinting of PGRP genes involves analyzing their DNA and protein sequences across a range of species and using statistical models to identify regions of the genes that are evolving under positive selection. These regions are thought to be important for the recognition of bacterial pathogens and for mounting an effective immune response (Ahmad et al., 2020). Our Study using evolutionary fingerprinting have identified several regions of PGRP genes that are evolving rapidly and are likely to be under positive selection. These regions are often located in the peptidoglycan-binding domain of PGRPs, which is responsible for recognizing bacterial cell wall components. In addition to identifying positively selected regions, evolutionary fingerprinting can also be used to study the functional consequences of genetic variation in PGRP genes. For example, studies have shown that mutations in PGRP genes can affect the sensitivity and specificity of the immune response, which can influence susceptibility to infectious diseases (Eleftherianos and Castillo, 2012)..
We discovered that these proteins had undergone adaptive evolution, with varying replacement rates at various places in their amino acid sequences. For PGLYRP, the average substitution rate was roughly 1.38, 0.54, and 0.12, respectively, based on various ratio groupings, with the least being 0.12 for various amino acid sites in PGLYRP (Fig. 3). Several studies have used these parameters to investigate the evolutionary dynamics of PGRP proteins in Drosophila and other organisms.For example, a study by Wang (2019) used Stanfel class parameters to estimate the amino acid substitution rates in PGRP proteins across different insect species. They found that the substitution rates varied among different PGRP genes and among different regions of the same protein, suggesting differences in the functional constraints acting on these regions (Wang et al., 2019). We found that the average amino acid substitution rates for PGRP proteins were roughly 1.38, 0.54, and 0.12, depending on the specific PGRP gene and the organism being analyzed. Furthermore, they discovered a function for adaptive evolution in directing the evolution of these immune-related proteins by finding evidence of positive selection operating on certain amino acid residues in several PGRP genes.The amino acids were divided into three groups through the evolutionary rate cluster: those in which the substitution pair FWY and HKR accounted for roughly fifty percent of the total, those in which DENQ accounted for fifty percent, and those in which ACGILMPSTV traded ninety percent. The evolution of PGRP protein in Drosophila involves changes in amino acid substitution rates, which can provide insights into the functional and evolutionary dynamics of this protein. Several studies have investigated the patterns of amino acid substitution in the PGRP protein family across different Drosophila species. For example, a study by Biswas et al. (2014) found that the PGRP family shows a higher rate of amino acid substitution than other immune-related proteins in Drosophila. They suggested that this high rate of substitution is due to the rapid evolution of PGRP proteins, which is likely driven by host-pathogen coevolution (Biswas et al., 2014). It was found that PGRP genes showed variable rates of amino acid substitution, with some genes evolving more rapidly than others and suggested that these differences in evolutionary rates may be due to differences in the functional constraints acting on these genes. Overall, evolutionary fingerprinting of PGRP genes provides insights into the molecular evolution of the innate immune system and the mechanisms by which host organisms adapt to new and emerging bacterial pathogens. This approach may have important implications for the development of novel strategies for the treatment and prevention of infectious diseases.
5 Conclusion
In conclusion, this study sheds light on the complex molecular evolution of PGN recognition proteins and their role in regulating the immune response against infectious diseases in D. melanogaster. The research reveals that PGN recognition proteins have undergone rapid and diverse evolution, with some experiencing positive selection and others undergoing gene duplication and loss. Furthermore, the study shows that different PGN proteins play distinct roles in regulating the immune response to bacterial infections, with some responding specifically to certain types of bacteria. These findings have important implications for understanding the mechanisms of pathogen recognition and immune defense in animals and the potential for developing new treatments for infectious diseases. Overall, this study provides valuable insights into the evolution and function of PGN recognition proteins in the immune response of D. melanogaster and highlights their potential relevance to other organisms. The molecular evolution of pathogen recognition peptidoglycan proteins in D. melanogaster is a complex and dynamic process that plays a crucial role in regulating the immune response against infectious diseases. The genetic and molecular information available for D. melanogaster is limited, which can hinder the accuracy and comprehensiveness of the study. Further research in this area will undoubtedly provide a more comprehensive understanding of the mechanisms underlying animal pathogen recognition and immune defense. The study may not have investigated the functional implications of the molecular evolution of PGRPs in D. melanogaster, which can limit our understanding of the role of these proteins in regulating the immune response against infectious diseases.
Acknowledgment
The authors extend their appreciation to the Researchers Supporting Project number (RSP2023R165), King Saud University, Riyadh, Saudi Arabia.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Adaptive evolution of peptidoglycan recognition protein family regulates the innate signaling against microbial pathogens in vertebrates. Microb. Pathog.. 2020;147:104361.
- [Google Scholar]
- Positive selection drives the adaptive evolution of mitochondrial antiviral signaling (MAVS) proteins-mediating innate immunity in mammals. Front. Veterin. Sci.. 2022;8:1680.
- [Google Scholar]
- Ahmad, H.I., Afzal, G., Sadia, S., et al., 2022. Structural and evolutionary adaptations of Nei-like DNA glycosylases proteins involved in base excision repair of oxidative DNA damage in vertebrates. Oxidative medicine and cellular longevity. 2022.
- Molecular Evolution of the Bactericidal/Permeability-Increasing Protein (BPIFA1) Regulating the Innate Immune Responses in Mammals. Genes. 2022;14(1):15.
- [Google Scholar]
- ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res.. 2016;44(W1):W344-W350.
- [Google Scholar]
- Natural selections: selfish altruists, honest liars, and other realities of evolution. Bellevue Literary Press; 2007.
- Inference of episodic changes in natural selection acting on protein coding sequences via CODEML. Curr. Protocols Bioinformat.. 2016;54(1) 6.15.11-16.15.32
- [Google Scholar]
- Evolutionary multiobjective optimization in dynamic environments: A set of novel benchmark functions. In: 2014 IEEE Congress on Evolutionary Computation (CEC). IEEE; 2014.
- [Google Scholar]
- Protein Data Bank (PDB): the single global macromolecular structure archive. Protein Crystall.: Methods Protocols. 2017:627-641.
- [Google Scholar]
- ConSurf: using evolutionary data to raise testable hypotheses about protein function. Isr. J. Chem.. 2013;53(3–4):199-206.
- [Google Scholar]
- Evolution of reproductive proteins from animals and plants. Reproduction. 2006;131(1):11-22.
- [Google Scholar]
- Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr.. 2002;40(1):82-92.
- [Google Scholar]
- Mechanisms and biological impacts of graphene and multi-walled carbon nanotubes on Drosophila melanogaster: Oxidative stress, genotoxic damage, phenotypic variations, locomotor behavior, parasitoid resistance, and cellular immune response. J. Appl. Toxicol.. 2022;42(3):450-474.
- [Google Scholar]
- Investigating and characterizing the direct binding interactions between chemotherapeutic cisplatin and pattern recognition receptor. Toll-like Receptor. 2022;4
- [Google Scholar]
- MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinf.. 2004;5(1):1-19.
- [Google Scholar]
- Molecular mechanisms of aging and immune system regulation in Drosophila. Int. J. Mol. Sci.. 2012;13(8):9826-9844.
- [Google Scholar]
- Detecting balancing selection in genomes: limits and prospects. Mol. Ecol.. 2015;24(14):3529-3545.
- [Google Scholar]
- A simple model host for identifying Gram-positive virulence factors. Proc. Nat. Acad. Sci.. 2001;98(19):10892-10897.
- [Google Scholar]
- The Drosophila speciation factor HMR localizes to genomic insulator sites. PLoS One.. 2017;12(2):e0171798.
- [Google Scholar]
- ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics. 2003;19(1):163-164.
- [Google Scholar]
- The ConSurf-DB: pre-calculated evolutionary conservation profiles of protein structures. Nucleic Acids Res.. 2009;37(suppl_1):D323-D327.
- [Google Scholar]
- A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol.. 2003;52(5):696-704.
- [Google Scholar]
- Hall, T., 2004. BioEdit version 7.0. 0. Distributed by the author, website: http://www.mbio.ncsu.edu/BioEdit/bioedit.html.
- Genome-Wide Analysis of Gene Families of Pattern Recognition Receptors in Fig Wasps (Hymenoptera, Chalcidoidea) Genes. 2021;12(12):1952.
- [Google Scholar]
- Maximum likelihood of phylogenetic networks. Bioinformatics. 2006;22(21):2604-2611.
- [Google Scholar]
- Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol.. 2006;23(10):1891-1901.
- [Google Scholar]
- Peptidoglycan recognition proteins in Drosophila immunity. Dev. Comp. Immunol.. 2014;42(1):36-41.
- [Google Scholar]
- LigPlot+: Multiple Ligand–protein Interaction Diagrams for Drug Discovery. ACS Publications; 2011.
- A Population and Quantitative Genetic analysis of the Drosophila Melanogaster Antibacterial Immune Response. The Pennsylvania State University; 2002.
- Natural selection on the Drosophila antimicrobial immune system. Curr. Opin. Microbiol.. 2008;11(3):284-289.
- [Google Scholar]
- The rapid evolution of signal peptides is mainly caused by relaxed selection on non-synonymous and synonymous sites. Gene. 2009;436(1–2):8-11.
- [Google Scholar]
- Peptidoglycan recognition protein SC (PGRP-SC) shapes gut microbiota richness, diversity and composition by modulating immunity in the house fly Musca domestica. Insect Mol. Biol. 2022
- [Google Scholar]
- Pattern recognition receptors in Drosophila immune responses. Dev. Comp. Immunol.. 2020;102:103468.
- [Google Scholar]
- Towards realistic codon models: among site variability and dependency of synonymous and non-synonymous rates. Bioinformatics. 2007;23(13):i319-i327.
- [Google Scholar]
- Evolutionary origin of peptidoglycan recognition proteins in vertebrate innate immune system. BMC Evol. Biol.. 2011;11(1):1-10.
- [Google Scholar]
- gcodeml: a Grid-Enabled Tool for Detecting Positive Selection in Biological Evolution. HealthGrid; 2012.
- Detecting individual sites subject to episodic diversifying selection. PLoS genetics.. 2012;8(7):e1002764.
- [Google Scholar]
- FUBAR: a fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol.. 2013;30(5):1196-1205.
- [Google Scholar]
- Predictive modelling of angiotensin converting enzyme inhibitory dipeptides. Food Chem.. 2012;133(4):1349-1354.
- [Google Scholar]
- Quantifying adaptive evolution in the Drosophila immune system. PLoS Genet.. 2009;5(10):e1000698.
- [Google Scholar]
- The whole genome sequence of the Mediterranean fruit fly, Ceratitis capitata (Wiedemann), reveals insights into the biology and adaptive evolution of a highly invasive pest species. Genome Biol.. 2016;17:1-31.
- [Google Scholar]
- Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics. 2005;21(10):2531-2533.
- [Google Scholar]
- Innate recognition of intracellular pathogens: detection and activation of the first line of defense. APMIS.. 2009;117(5–6):323-337.
- [Google Scholar]
- Molecular evolution of the major arthropod chemoreceptor gene families. Annu. Rev. Entomol.. 2019;64:227-242.
- [Google Scholar]
- A globin gene of ancient evolutionary origin in lower vertebrates: evidence for two distinct globin families in animals. Mol. Biol. Evol.. 2005;22(1):12-20.
- [Google Scholar]
- Empirical evaluation of genetic clustering methods using multilocus genotypes from 20 chicken breeds. Genetics. 2001;159(2):699-713.
- [Google Scholar]
- Detection of protein three-dimensional side-chain patterns: new examples of convergent evolution. J. Mol. Biol.. 1998;279(5):1211-1227.
- [Google Scholar]
- Dynamic evolution of the innate immune system in Drosophila. Nat. Genet.. 2007;39(12):1461-1468.
- [Google Scholar]
- Toll-mediated Cellular Immune Response in Drosophila Melanogaster. Umeå universitet; 2014.
- Evolution and function of thioester-containing proteins and the complement system in the innate immune response. Front. Immunol.. 2017;8:759.
- [Google Scholar]
- Peptidoglycan recognition proteins: on and off switches for innate immunity. Immunol. Rev.. 2004;198(1):83-96.
- [Google Scholar]
- Selecton 2007: advanced models for detecting positive and purifying selection using a Bayesian inference approach. Nucleic Acids Res.. 2007;35(suppl_2):W506-W511.
- [Google Scholar]
- MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol.. 2013;30(12):2725-2729.
- [Google Scholar]
- Toll and IMD pathways synergistically activate an innate immune response in Drosophila melanogaster. Mol. Cell. Biol.. 2007;27(12):4578-4588.
- [Google Scholar]
- Regulators of the Toll and Imd pathways in the Drosophila innate immune response. Trends Immunol.. 2005;26(4):193-198.
- [Google Scholar]
- Duplication and divergence: the evolution of new genes and old ideas. Annu. Rev. Genet.. 2004;38:615-643.
- [Google Scholar]
- FlyBase: enhancing Drosophila gene ontology annotations. Nucleic Acids Res.. 2009;37(suppl_1):D555-D559.
- [Google Scholar]
- Peptidoglycan recognition proteins in insect immunity. Mol. Immunol.. 2019;106:69-76.
- [Google Scholar]
- SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res.. 2018;46(W1):W296-W303.
- [Google Scholar]
- Datamonkey 2.0: a modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol.. 2018;35(3):773-777.
- [Google Scholar]
- Comparative genomics approaches to study organism similarities and differences. J. Biomed. Inform.. 2002;35(2):142-150.
- [Google Scholar]
- The Kyoto encyclopedia of genes and genomes–KEGG. Yeast (Chichester, England).. 2000;17(1):48-55.
- [Google Scholar]
- Comparison and Phylogenetic Analysis of Mitochondrial Genomes of Talpidae Animals. Animals.. 2023;13(2):186.
- [Google Scholar]
- PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol.. 2007;24(8):1586-1591.
- [Google Scholar]
- The Drosophila amidase PGRP-LB modulates the immune response to bacterial infection. Immunity. 2006;24(4):463-473.
- [Google Scholar]
- Prediction of protein-protein interaction sites using constructive neural network ensemble. In: 2010 International Conference on Computational Intelligence and Software Engineering. IEEE; 2010.
- [Google Scholar]
- Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell Reports Methods.. 2021;1(3):100014.
- [Google Scholar]




