

Translate this page into:
Molecular docking and molecular dynamics simulation of anticancer active ligand ‘3,5,7,3′,5′-pentahydroxy-flavanonol-3-O-α-L-rhamnopyranoside’ from Bauhinia strychnifolia Craib to the cyclin-dependent protein kinase
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The compound ‘3,5,7,3′,5′-pentahydroxy-flavanonol-3-O-α-L-rhamnopyranoside’ reported from Bauhinia strychnifolia Craib (family Fabaceae) possess ten times more cytotoxicity against certain cancer cell line than the anti-cancer drugs, but nontoxic to normal cells. Its stability with the protein ‘cyclin-dependent protein kinase 2/CDK-2′ (-which plays vital role in apoptosis, regulation of the cell cycle, transcription and in the neuronal functions) were performed via molecular docking and molecular dynamics simulation simulations. The molecular dynamics simulation suggests that the protein-ligand complex is stable at least in the time period of 40 ns. This study also shows that after 20 ns of simulation, there is a shift in the ligand position, and it might be due to changes in the binding type. According to the data obtained via MM-PBSA calculations, this shift leads to even stronger binding of ligand. Such binding flexibility can be explained by highly hydrophilic nature of both ligand and binding site.
Keywords
Molecular docking
Molecular docking simulation
3,5,7,3′,5′-pentahydroxy-flavanonol-3-O-α-L-rhamnopyranoside
Bauhinia strychnifolia Craib
Fabaceae
Cyclin-dependent protein kinase 2

1 Introduction
Despite remarkable decline in death rates owing to cancer during last two to three decades, it still remains world’s major health issue, and the leading causes of death worldwide too (Siegel et al., 2016). Apart from the strategies such as targeted cancer therapy (Padma, 2015), immunotherapy (Upadhyaya, 2006), hyperthermia (Wust et al., 2002), resistance modulation (Fracasso et al., 2000), antidotes/toxicity modifiers (Hasinoff et al., 1999) and alternative formulations (Krishna and Mayer, 1997), the surgery, chemotherapy and the radiotherapy are still the main tones in the treatment of cancer, but it brings severe side effects too (DeSantis et al., 2014). Recently Several novel anticancer candidates such as natural product (Zengin et al., 2018; Trampetti et al., 2019; Uysal et al., 2019), organic synthetic compound (Mollica et al., 2011, 2012; Schreiber, 2011; Nicolaou, 2014; Picot et al., 2017) and nanoparticles (Brigger et al., 2002) have been identified recently; however, still the natural products used as an effective drug candidate for inhibiting cancer (Thenmozhi et al., 2018), and are continuously being explored through plant bioprospecting because of its safer nature compared to the synthetic drug molecules (Ali et al., 2014; Newman and Cragg, 2016).
Bauhinia strychnifolia Craib (family Fabaceae) traditionally used for the treatment of the cancer (Wutthithammavet, 1997; Yuenyongsawad et al., 2013). The compound ‘3,5,7,3′,5′-pentahydroxy-flavanonol-3-O-α-L-rhamnopyranoside’ isolated from the ethanolic extract of root and stem of B. strychnifolia have been reported ten times more cytotoxic (IC50: 0.00054 μg/mL) against KB cell line than camptothecin (-the anti-cancer drug, IC50: 0.0057 μg/mL), while nontoxic to normal cells (Yuenyongsawad et al., 2013). The recent advances in molecular docking and molecular dynamics simulation have provided insights of the compounds including mechanism of action, drug-like properties, selectivity, improved potency and side effects (Kitchen et al., 2004). The protein CDK-2 exerts catalytic functions when bound with the cyclins, and plays vital role in apoptosis, regulation of the cell cycle, transcription and in the neuronal functions (Dai and Grant, 2003; Huwe et al., 2003). Hence, in the present molecular docking and molecular dynamics simulation simulations aimed to evaluate the binding of the ligand with the core-cell cycle machinery protein.
2 Materials and methods
The 2D structure of ligand ‘3,5,7,3′,5′-pentahydroxy-flavanonol-3-O-α-L-rhamnopyranoside’ reported to have anticancer activity (Yuenyongsawad et al., 2013) was modeled, converted to 3D structure (using ChemSketch v. 12.01), optimized using MMFF force field (parameters: 500 steps of steepest descent algorithm, convergence criterion of 10e-7) (Halgren, 1996), and used for docking simulation (Kumari et al., 2014) with receptor CDK-2 (PDB ID: 1DI8) retrieved from Protein Data Bank. During the preparation of receptors, all heteroatoms (i.e. water, ions, etc.) were removed, polar hydrogens were added, and Kollman charges were assigned (Case et al., 2016; da Silva and Vranken, 2012). The ligand was docked using AutoDock4.2 (Trott and Olson, 2010), the trajectories of docked complexes were analyzed through 20 ns of MD simulations using GROMACS 4.6.5 (Hess et al., 2008) following the previously mentioned procedure (Phosrithong and Ungwitayatorn, 2010; Sudhamalla et al., 2010; Gurung et al., 2016; Das et al., 2018a,b).
3 Results and discussion
The molecular docking of the known ligand (DTQ – 4-[3-hydroxyanilino]-6,7-dimethoxyquinazoline) was carried out into CDK-2 complex structure. The RMSD for the position of the atoms was selected as a quantitative measure of quality. In this case, it was about 1.5 Å. The procedure was performed using several molecular docking software approaches for CDK-2. The program resulted in lowest RMSD in comparison to X-ray structure was selected (Table 1). The Fig. 1A shows superposition of docked ligand with ligand in X-ray structure.
S. No.
Docking software
CDK-2 (PDB code 1DI8)
1.
Autodock Vina
1.33
2.
AutoDockFR
3.49
3.
PharmDock
3.75
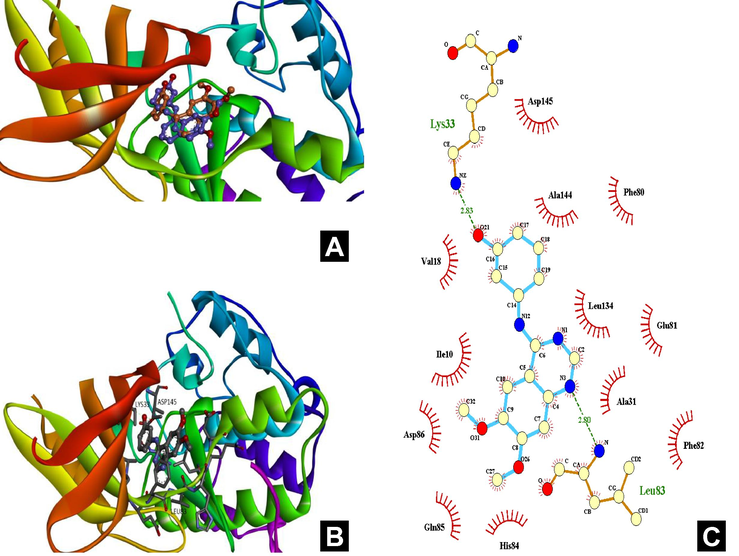
A. Superposition of ligand in the protein-ligand complex, obtained by docking and using X-ray data. Orange – ligand from the pdb file, purple – position of this ligand calculated by docking. Fig. 1B-C. General view and position of active site.
3.1 Binding site
As the typical protein kinase CDK-2 formed by two flexible fragments: N and C-lobes, between them the most studied site-ATP-competitive binding site is placed. This site has a high degree of conservation among the cyclin-dependent protein kinases and in general it tends to bind to polar heteroaromatic compounds (Sims et al., 2003). Various inhibitors normally occupy three different sub-sites (the adenine, ribose and triphosphate subsites) or three specific binding modes for ATP competitive inhibitors (Li et al., 2015). The first one is observed for compounds with a heterocyclic purine moiety. Such compounds inhibit CDK-2 by occupation preferably adenine sub-site and in general replicate the location of ATP in the binding site. H-bonds formation with the backbones of residues Glu81 and Leu83 is crucial for this binding mode. Another type of binding observed in 4-anilinoquinazoline inhibitor class. Such compounds form multiple bridged hydrogen bonds with water, also characterized by pi-pi staking with PHE80. Inhibitors with flavonoid structure provide the third way of binding. Two of the conserved H-bonds with Leu83 and Glu81 can be observed in this binding mode, additionally residues Lys33 and Asp145 which are located in the ribose and triphosphate regions of ATP-competitive binding site respectively also take part in H-bond formation with the ligand. Ligand, which position is shown in the complex with CDK-2, binds in the way that is typical for the flavonoids. The Fig. 1B-C shows key interactions of this ligand with amino acids of the binding site.
3.2 Binding interaction
The 3D model of interaction of the most favored docking pose in CDK-2 (Fig. 2A) show typical flavonoid type of binding interaction of phenolic hydroxyl groups with the side chains of Asp145 and Lys33. In addition, there was a formation of hydrogen bonds between the hydroxyl group of the carbohydrate moiety and Glu12. There might be possibilities for hydrogen bonds between ligand hydroxyl groups and water molecules which occupy the binding site too. The pharmacophore model was constructed on the basis of docking data (coordinates of the ligand). This model taken into account for the key interaction established in the result of analysis of the ligand position into the binding site. The H-bonds model is shown in the Fig. 2B.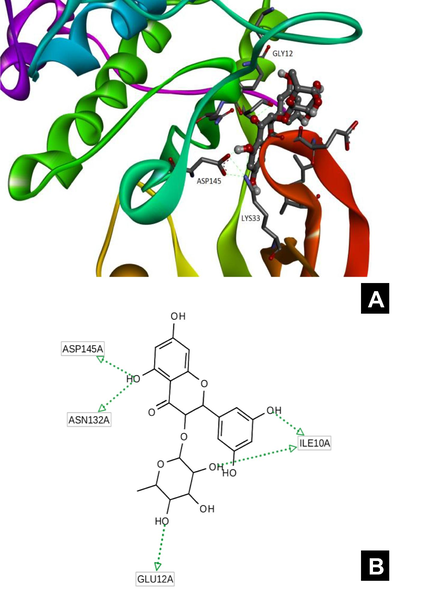
A-B. Protein-ligand complex with target compound in the binding site. A (left) – general view of docking pose. B (right) – h-bond map. Different colors of the protein show different secondary structure elements. Residues of the protein are highlighted by blue color. In the ligand the red atoms represent oxygen, white atoms – hydrogen, yellow – sulfur, dark blue – nitrogen and grey – carbon.
3.3 Possible modifications of inhibitor’s structure
The ligand showed the ability to form a large number of hydrogen bonds with the amino acids of binding site and bridge hydrogen bonds with water molecules. This is reflected in the high values of docking scoring functions, which prompts to assume that the affinity of the ligand to the ATP-competitive binding site of CDK-2 will be significantly higher. However, when it comes to protein kinases with high degree of conservation of the binding sites, and compound of interest belongs to the class of flavonoids which are well known by their ability to inhibit different kinases, the issue of selectivity comes into picture.
3.4 Analysis of the trajectory and MM-PBSA analysis
The Fig. 3A shows the RMSD values of the protein-ligand complex. The substantial RMSD value can be explained by the mobility of the loops. At the same time, the visual analysis of the trajectory confirms the stability of the secondary structure of the protein. There was a significant shift in RMSD values for ligand after 20 ns of simulation. The RMSD rises from an average of 0.10 nm with SD of 0.01 nm to 0.19 nm with SD of 0.02 nm. This shift may indicate a new binding mode for ligand. The MM-PBSA calculations were performed to explore the influence of this event on binding energy. The Fig. 3B shows binding energy – time dependence during simulation. The significant change of the average values of binding energy (from −28.7 kJ/ mol to −50.9 kJ/ mol) in about the same time when the shift of RMSD values has been occurred. Increased binding energy shows that the new type of binding can be more energetically favorable.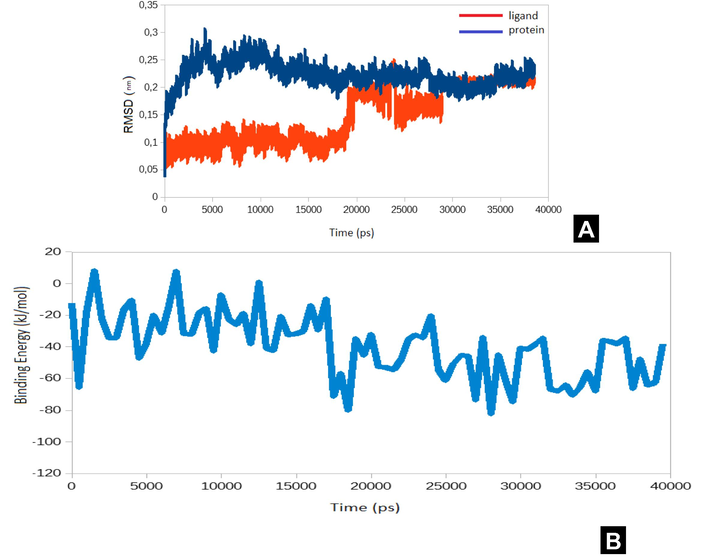
A. The RMSD values of the protein-ligand complexes over the simulation time (X-axis represents time scale in nanosecond and Y-axis represent the RMSD in nanometers). B. Average binding energy. One can see the significant change of the average values of binding energy (from −28.7 kJ/mol to −50.9 kJ/mol) in about the same time when the shift of RMSD values has been occurred. Increased binding energy shows that the new type of binding can be more energetically favorable. Following is the more detailed analysis of a new type of binding.
3.5 Analysis of the conformational changes
Several representative frames from the part of trajectory after 30 ns has been studied for the analysis of a new type of binding. One can observe the change of the position of the carbohydrate moiety as well as changes in the conformation of the benzene ring. This position of the ligand enables to form an additional hydrogen bonds with the Glu12. The detailed picture of this interactions have been shown in the Fig. 4A-B.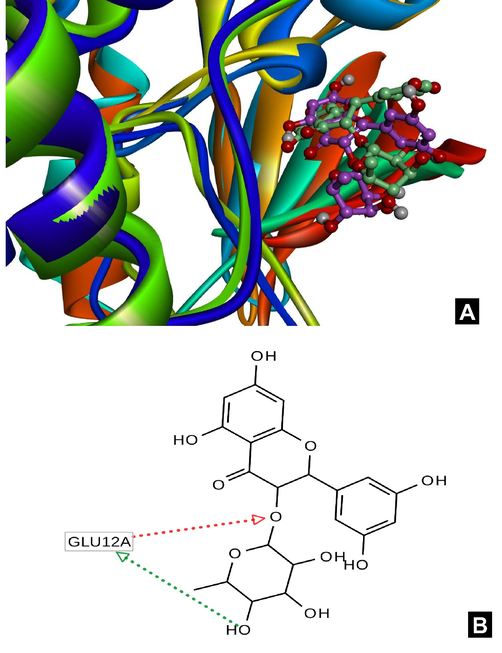
A-B. A. Superposition of protein-ligand complex (green color for ligand) with target compound in the binding site obtained by docking and extracted from molecular dynamics trajectory (magenta color for ligand). B. h-bond map. Different colors of the protein show different secondary structure elements.
4 Conclusions
The binding study shows that the flavonoid type of binding is the most probable binding mode for the ligand. The molecular dynamics simulation suggests that protein-ligand complex is stable at least in the time period of 40 ns. This study also show that after 20 ns of simulation, there is a shift in ligand position, and it might be due to binding type change. According to the data obtained via MM-PBSA calculations, this shift leads to even stronger binding of ligand. Such binding flexibility can be explained by highly hydrophilic nature of both ligand and binding site. Despite that molecular dynamics simulation studies show good chances for actual binding, it is important to note that the issue of selectivity is very actual for flavonoids and as a consequence for ligand.
Acknowledgments
Research supported by the King Saud University, Deanship of Scientific Research, College of Science, Research Center.
References
- In vitro cytotoxicity screening of some wild plants extracts from Saudi Arabia on human breast adenocarcinoma cells. Genet. Mol. Res.. 2014;13(2):3981-3990.
- [Google Scholar]
- Nanoparticles in cancer therapy and diagnosis. Adv. Drug Deliv. Rev.. 2002;54(5):631-651.
- [Google Scholar]
- AMBER 2016. San Francisco: University of California; 2016.
- Molecular recognition of bio-active flavonoids quercetin and rutin by bovine hemoglobin: an overview of the binding mechanism, thermodynamics and structural aspects through multi-spectroscopic and molecular dynamics simulation studies. Phys. Chem. Chem. Phys.. 2018;20(33):21668-21684.
- [Google Scholar]
- Deciphering the interaction of 5,7-dihydroxyflavone with hen-egg-white lysozyme through multispectroscopic and molecular dynamics simulation approaches. ChemistrySelect. 2018;3(17):4911-4922.
- [Google Scholar]
- Cancer treatment and survivorship statistics, 2014. CA Cancer J. Clin.. 2014;64(4):252-271.
- [Google Scholar]
- Phase I study of paclitaxel in combination with a multidrug resistance modulator, PSC 833 (Valspodar), in refractory malignancies. J. Clin. Oncol.. 2000;18:1124-1134.
- [Google Scholar]
- Exploring the physicochemical profile and the binding patterns of selected novel anticancer Himalayan plant derived active compounds with macromolecular targets. Inf. Med. Unlocked. 2016;5:1-14.
- [Google Scholar]
- Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem.. 1996;17:490-519.
- [Google Scholar]
- The cardioprotective and DNA topoisomerase II inhibitory agent dexrazoxane (ICRF-187) antagonizes camptothecin-mediated growth inhibition of Chinese hamster ovary cells by inhibition of DNA synthesis. Anticancer Drugs. 1999;10:47-54.
- [Google Scholar]
- GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput.. 2008;4:435-447.
- [Google Scholar]
- Small molecules as inhibitors of cyclin-dependent kinases. Angew. Chem. Int. Ed. Engl.. 2003;42:2122-2138.
- [Google Scholar]
- Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discov.. 2004;3(11):935-949.
- [Google Scholar]
- Liposomal doxorubicin circumvents PSC 833-free drug interactions, resulting in effective therapy of multidrug-resistant solid tumors. Cancer Res.. 1997;57:5246-5253.
- [Google Scholar]
- Kumari, R., Kumar, R., Open Source Drug Discovery Consortium, Lynn A., 2014. g_mmpbsa – a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 54(7), 1951–1962.
- Insights on Structural characteristics and ligand binding mechanisms of CDK2. Int. J. Mol. Sci.. 2015;16:9314-9340.
- [Google Scholar]
- Synthesis of (S)-5,6-dibromo-tryptophan derivatives as building blocks for peptide chemistry. Tetrahedron Lett.. 2011;52(20):2583-2585.
- [Google Scholar]
- Synthesis and bioactivity of secondary metabolites from marine sponges containing dibrominated indolic systems. Molecules. 2012;17(5):6083-6099.
- [Google Scholar]
- Organic synthesis: the art and science of replicating the molecules of living nature and creating others like them in the laboratory. Proc. R. Soc. A. 2014;470:20130690.
- [Google Scholar]
- Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod.. 2016;79(3):629-661.
- [Google Scholar]
- Padma, V.V., 2015. An overview of targeted cancer therapy. 5(4), 1–6.
- Molecular docking study on anticancer activity of plant-derived natural products. Med. Chem. Res.. 2010;19:817-835.
- [Google Scholar]
- In vitro and in silico studies of mangiferin from Aphloia theiformis on key enzymes linked to Diabetes type 2 and associated complications. Med. Chem.. 2017;13(7):633-640.
- [Google Scholar]
- Organic synthesis toward small-molecule probes and drugs. PNAS. 2011;108(17):6699-6702.
- [Google Scholar]
- A computational model of binding thermodynamics: the design of cyclin-dependent kinase 2 inhibitors. J. Med. Chem.. 2003;46(15):3314-3325.
- [Google Scholar]
- Molecular dynamics simulation and binding studies of β-Sitosterol with human serum albumin and its biological relevance. J. Phys. Chem. B. 2010;114(27):9054-9062.
- [Google Scholar]
- Pharmacological credence of the folklore use of Bauhinia malabarica in the management of jaundice. Saudi J. Biol. Sci.. 2018;25:22-26.
- [Google Scholar]
- Exploring the halophyte Cistanche phelypaea (L.) Cout as a source of health promoting products: in vitro antioxidant and enzyme inhibitory properties, metabolomic profile and computational studies. J. Pharm. Biomed. Anal.. 2019;165(20):119-128.
- [Google Scholar]
- AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem.. 2010;31(2):455-461.
- [Google Scholar]
- Upadhyaya, S.K., 2006. Immunotherapy for cancer with special reference to lung cancer, Apollo Med., 3, 277–284.
- Multifunctional approaches to provide potential pharmacophores for the pharmacy shelf: Heracleum sphondylium L. subsp. ternatum (Velen.) Brummitt. Comput. Biol. Chem.. 2019;78:64-73.
- [Google Scholar]
- Thai Traditional Medicine. Bangkok: Odean Store Press; 1997. p. :381.
- Anti-cancer activity of compounds from Bauhinia strychnifolia stem. J. Ethnopharmacol.. 2013;150(2):765-769.
- [Google Scholar]
- Combination of phenolic profiles, pharmacological properties and in silico studies to provide new insights on Silene salsuginea from Turkey. Comput. Biol. Chem.. 2018;77:178-186.
- [Google Scholar]




