

Translate this page into:
Molecular docking and dynamics studies on propolis sulabiroin-A as a potential inhibitor of SARS-CoV-2
⁎Corresponding author. jakafajar@ui.ac.id (Jaka Fajar Fatriansyah),
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Molecular docking and dynamics simulations were conducted to investigate the antiviral activity of Propolis Sulabiroin-A to inhibit the SARS-CoV-2 virus with quercetin, hesperidin, and remdesivir as control ligands. The parameters calculated were docking score and binding energy/molecular mechanics-generalized born surface area (MMGBSA), root mean square displacement (RMSD), and root mean square fluctuation (RMSF). Docking and MMGBSA scores showed that all the ligands demonstrate an excellent candidate as an inhibitor, and the order of both scores is hesperidin, remdesivir, quercetin, and sulabiroin-A. The molecular dynamics simulation showed that all the ligands are good candidates as inhibitors. Although the fluctuation of Sulabiroin-A is relatively high, it has less protein–ligand interaction time than other ligands. Overall, there is still a good possibility that sulabiroin-A can be used as an alternative inhibitor if a new structure of receptor SARS-CoV-2 is used.
Keywords
SARS-CoV-2
Molecular docking
Molecular dynamics
Drug discovery
Sulabiroin-A

- COVID-19
-
Corona Virus Disease 2019
- MD
-
Molecular Dynamic
- MM-GBSA
-
Molecular Mechanics/Generalized Born Surface Area
- RMSD
-
Root Mean Square Deviation
- RMSF
-
Root Mean Square Fluctuation
- SARS-Cov-2
-
Severe acute respiratory syndrome coronavirus 2
Abbreviations
1 Introduction
Coronavirus disease 2019 (COVID-19) started in the Wuhan People's Republic of China in December 2019. It has spread rapidly to almost every country, with 209,201,939 confirmed cases and 4,390,467 confirmed deaths worldwide (World Health Organization Data reported on 19 August 2021 6:00 CEST). The USA, Brazil, and India are the initially hardest-hit countries, followed by Russia, South Africa, Mexico, Peru, and Indonesia. Thus, the availability of SARS-CoV-2 virus inhibitors is urgently needed.
Various efforts have been made to discover drugs against the virus SARS-CoV-2. Antiviral for Ebola, remdesivir, has been reported to effectively inhibit SARS-CoV-2 virus infection in vitro along with hydroxychloroquine (Liu et al., 2020). In addition to in vitro investigations, chloroquine (Gao et al., 2019), oseltamivir (Muralidharan et al., 2020), and lopinavir/ritonavir (Cao et al., 2020; Chang et al., 2020) have been investigated computationally in an effort to find the most effective drug for fighting against this virus.
SARS-CoV-2 is a beta-coronavirus that belongs to the subgenus sarbecovirus, Ortho Coronaviridae subfamily (Zhu et al., 2020). The genome sequence of SARS-CoV-2 is 96.2%, similar to bat CoV RaTG13, and 79.5% similar to SARS-CoV (Guo et al., 2019). SARS-CoV-2 uses angiotensin-converting enzyme 2 (ACE2), which is the same receptor as SARS-CoV, to infect humans (Zhou et al., 2020).
Sulabiroin-A is a compound that can be isolated from the propolis of stingless bees (Tetragonula aff. biroi) collected in South Sulawesi, Indonesia (Miyata et al., 2020). Propolis is a well-known medicinal beehive product due to its antiviral properties (Kwon et al., 2020). Propolis was reported to have beneficial biological activity, including antiviral antibiotics, antifungal, antiviral, and antimicrobial activities (Burdock et al., 1998; Kujumgiev et al., 1999; Miyata et al., 2020; Yildirim et al., 2016). Molecular docking studies have found that sulabiroin-A has SARS-CoV-2-inhibiting potency (Khayrani et al., 2020; Sahlan et al., 2021).
Indonesia is currently (April 2021) one of the fastest-growing coronavirus spreading countries, which strains its resources, including inadequate medical resources. The availability of SARS-CoV-2 antiviral agents is urgently needed. Furthermore, Indonesia has the availability of required natural resources, which can be extracted to synthesize Sulabiroin-A.
Molecular docking and dynamics are powerful computational methods to discover a new drug and have consequent real applicability in drug discovery (Salmaso and Moro, 2018). To computationally investigate a compound/drug candidate, we need control ligands. In our study, we used quercetin and hesperidin as control ligands under the classes of flavonoids and remdesivir. Flavonoids are known as antioxidants and antimicrobial compounds. Quercetin, a bioflavonoid, is widely used as an antiviral for several viruses, such as dengue virus, influenza virus, hepatitis C virus, and various viruses (Anusuya et al., 2017; Zandi et al., 2011). Quercetin is found in many plants, such as onion, guava, and cranberry. Hesperidin is a flavonoid compound with antimicrobial activities against human viruses despite its very low solubility in water (Abuelsaad, 2013; Liu et al., 2011; Yi et al., 2008). Hesperidin is found in the citrus family. Both compounds can be extracted from the various plants mentioned (Ko et al., 2011; Londoño-Londoño et al., 2010; Ma et al., 2008; Yang and Zhang, 2008).
On the other hand, in contrast to naturally extracted quercetin and hesperidin, remdesivir is a synthetic drug that can be synthesized in multiple steps from ribose derivatives. Remdesivir was initially developed by Gilead Sciences to treat Ebola virus disease and Marburg virus infections (Warren et al., 2016). In addition, remdesivir was found to show antiviral activity against other single-stranded RNA viruses, such as respiratory syncytial virus, Junin virus, Lassa fever virus, Nipah virus, Hendra virus, and coronaviruses (Lo et al., 2017, Mohapatra et al., 2021). In this study, we used remdesivir as a control ligand.
In this study, we explored molecular docking and molecular dynamics (MD) simulations to investigate the antiviral activity of sulabiroin-A with quercetin, hesperidin, and remdesivir as control ligands to inhibit the SARS-CoV-2 virus. In our molecular docking and dynamics study, the parameters calculated were docking score, binding energy, RMSD (root mean square displacement), and RMSF (root mean square fluctuation). The docking score and binding energy show the rating of the drugs relative to each other. RMSD (root mean square displacement) shows the stability of the docked complex. RMSF (root mean square fluctuation) indicates the conformational flexibility of the complex. The main objective of this paper is to provide insight into how these ligands can be used alternatively to fight the SARS-CoV-2 virus in strains of medical resources amid the coronavirus pandemic.
2 Materials and methods
2.1 Data collection
The structures of quercetin, hesperidin, remdesivir, and sulabiroin-A were drawn using MarvinSketch and saved as.mol files, which were then converted into.sdf files using Open Babel. The molecular structures are given in Fig. 1 (a), (b), (c), and (d) for quercetin, hesperidin, remdesivir, and sulabiroin-A, respectively.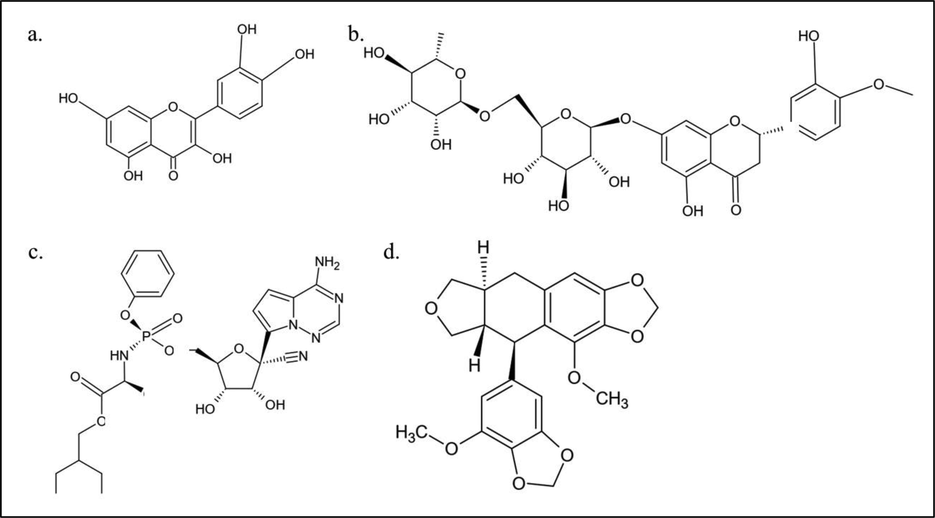
The molecular structure of 1 (a) quercetin, (b) hesperidin, (c) remdesivir and (d) sulabiroin-A.
2.2 Protein preparation
The structure of the SARS-CoV-2 main protease was retrieved from the Protein Data Bank (PDB ID: 6Y2F) (Zhang et al., 2020). Protease cleaves the polyprotein at approximately 11 (eleven) different sites. Proteases generate various nonstructural proteins that are important for viral replication and play a critical role in the replication of virus particles (Anand et al., 2003). This protease was used as a receptor. The protein was prepared in a suitable form using protein preparation wizard in Maestro ®, which has the following steps: (i) addition of missing disulfide bonds, (ii) removal of water molecules, (iii) addition of missing hydrogen atom (iv) fills missing side chain (vi) optimizing the hydrogen bonds to avoid steric clashes (vii) refining the structure with restrained minimization to an RMSD of 0.3 Å.
2.3 Ligand preparation
The ligands were prepared using Ligprep in Maestro. In the ligand preparation process, the ligands were desalted, and hydrogen atoms were added. All possible ionization and tautomeric states were prepared using the Epik configuration at pH 7 ± 2. The force fields used were OPLS3e, and the lowest penalty state was used for the subsequent processes.
2.4 Glide docking
Glide docking for the grid parameter using receptor grid generation in Maestro was set in the center of the inhibitor alpha-ketoamide in the protein's active site (Anusuya et al., 2017). The grid box coordinates were X = 11.476 Y = -1.396 Z = 20.745, with the size of the Box 20 Å. The van der Waals radius scaling was set to 1 to soften the potential for nonpolar parts of the receptor, and the partial charge was set to 0.25. All ligands were docked with the target protein with the extra precision (XP) algorithm. The advance setting was set so that a maximum of 5000 poses per ligand were generated during the initial docking phase, keeping at best 1000 poses per ligand for energy minimization, and 100 poses per ligand were filtered. The best-bound ligands to the active site were determined based on the most negative docking score.
2.5 Prime MM-GBSA calculation
The binding free energy (ΔG bind) of flavonoids and the drug target SARS-CoV-2 was calculated using the Prime MM-GBSA method. The best poses of protein–ligand complexes obtained in Glide XP docking were chosen to calculate the binding free energy (ΔG bind), as shown in Equation (1):
2.6 Molecular dynamics (MD) simulations
The docked ligand targets were merged in Maestro. Then, the system setup was used to model water TIP4P in a 10 Å orthorhombic box. The docked complex (merged ligand targets) was neutralized by the addition of ions automatically calculated by Maestro. Next, the equilibrated simulation time was set to 2 ns with a recording trajectory every 4.8 ps, and the ensemble class was assigned to NPT, with a temperature of 310 K. The obtained parameters were RMSD and RMSF.
3 Results and discussion
3.1 Molecular docking
Visual analyses were conducted by comparing the poses for each ligand-target interaction. Ligand interactions were already fitted in protein active sites. First, we discuss the analysis for Quercetin-target, in which its 2D projection interaction is shown in Fig. 2 (a). Three OH groups in quercetin form hydrogen bonds with Thr25, Asn142, and His41. In this case, quercetin acts as a donor for the hydrogen bond and, on the other hand, that amino acid acts as an acceptor.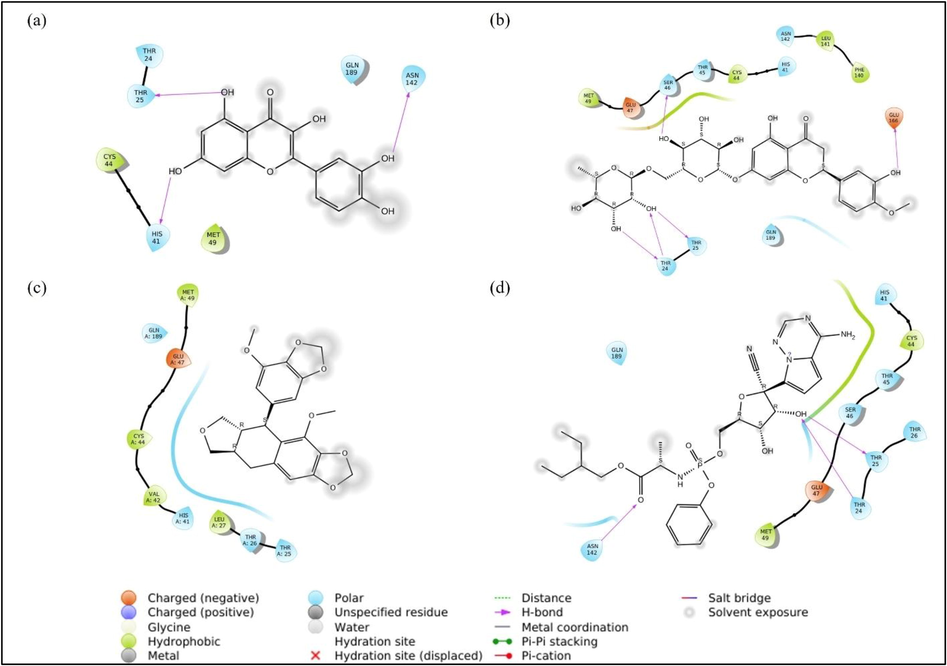
Ligand-target 2D projection of interaction (a) quercetin, (b) hesperidin, (c) sulabiroin-A, and (d) remdesivir.
Next, we discuss the hesperidin-target interaction, which is shown in Fig. 2 (a). The figure shows that hesperidin forms hydrogen bonds with the polar residues Thr24, Thr25, Ser46, and negatively charged Glu166. Here, Thr 24 shows interaction with two OH groups in the form of hydrogen bonds, where hesperidin acts as the donor and as an acceptor to the hydrogen bond with Thr 24. Here, the bonds formed are more complicated than those in the quercetin target, especially considering the complex interaction among Thr24, Thr25, and two OH groups in the hesperidin target. It can be estimated that the docking score for hesperidin targets will be more negative than that for quercetin targets.
Next, we discuss the Sulabiroin-A-target interaction, which is shown in Fig. 2 (c). Uniquely, the interaction does not indicate any bond between ligand and target, although the docking score is available. However, we observed that the ligands do not experience solvent exposure facing toward the target. This phenomenon may contribute to the docking score's negative value, although we estimate that the docking score is somewhat less negative than Quercetin-target and Hesperidin-target.
Next, we discuss the remdesivir-target interaction, which is shown in Fig. 2 (d). This indicates that the OH group forms a hydrogen bond with polar residues Thr24 and Thr25. This interaction is similar to the OH group interaction with Thr24 and Thr25 found in the hesperidin-ligand interaction. However, for the remdesivir-target case, only one OH group interacts, whereas the hesperidin-target case includes both OH groups at once. This phenomenon may suggest that the remdesivir-target docking score is less negative than the docking score for the hesperidin target.
In summary, the complexity of hydrogen bonds of ligand-target interactions indicates the strength of molecular docking. Quercetin has one hydrogen bond with Thr25, while hesperidin has more complex hydrogen bonds between Thr24 and Thr25. Sulabiroin-A has no bond, which indicates a weak interaction with the target. Remdesivir, like quercetin and hesperidin, has hydrogen bonds with Thr24 and Thr25, which is more complicated than quercetin but less complicated than hesperidin. Thus, from visual analyses, the best ligand to form a complex (ligand-target) is hesperidin-remdesivir-quercetin-sulabiroin-A, which was later approved by the ligand-target docking score and binding energy.
The scores of molecular dockings for quercetin, hesperidin, sulabiroin-A, and remdesevir are shown in Table 1. Docking results reveal that the hesperidin ligand has the lowest binding affinity toward the SARS-CoV-2 target. The lower value of the docking score shows that the ligand binding with the target is more stable.
No.
Ligand
Docking Score (kcal/mol)
Bind Energy(kcal/mol)
1
Quercetin
−6.704
−38.96
2
Hesperidin
−9.787
−52.07
3
Sulabiroin-A
−3.925
−30.97
4
Remdesivir
−7.511
−44.03
The differences between ligand-target interactions occurred due to the different structures bound to the target, resulting in the docking score, as shown in Table 1. The next step was to perform MMGBSA analysis for the binding energy of each ligand. MMGBSA consideration factors for the free binding energy are lipophilic, hydrogen bonds, electrostatic, covalent, van der Waals interactions, generalized Born electrostatic solvation energy, and π-π packing correction energy. Table 1 shows that hesperidin has the lowest free binding energy with a value of –52.07 kcal/mol, which shows that the ligand binds the substrate best among other ligands, followed by remdesivir at –44.03 kcal/mol. The binding energy of hesperidin and remdesivir was lower than that of quercetin, which verified that hesperidin and remdesivir have more robust binding compounds than quercetin.
Meanwhile, Sulabiroin-A shows only –30,97 kcal/mol to ensure that the binding is weakest among other ligands. The binding free energy result supported the role of MM-GBSA in distinguishing between real and decoy poses of a ligand in addition to rescoring datasets (Cavasotto, 2015). The value calculation was based on the influencing factor of MMGBSA shown in Fig. 3, and all the values were summed. All ligands’ primary contributions are van der Waals energy interactions followed by coulombic and lipophilic energy interactions. Only in Sulabiroin-A does the coulombic energy show a positive ΔG, which means that in Sulabiroin-A, electrostatic interactions are not considered ligand–protein interactions.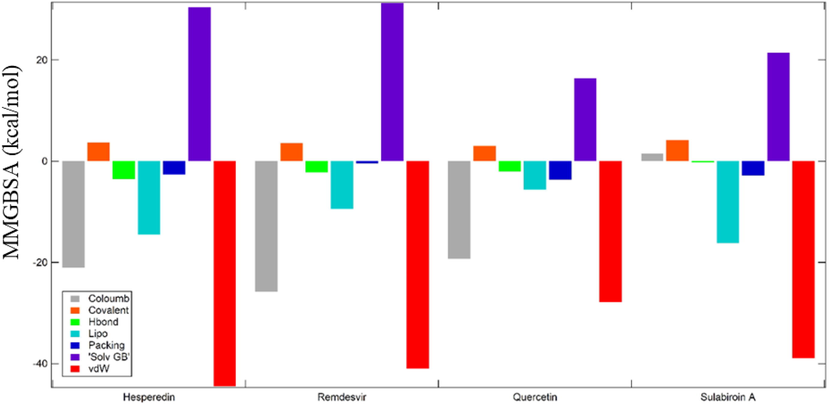
The contribution of various interactions to the MMGBSA score of ligands.
3.2 Molecular dynamics simulation
The stability of docked complexes and the binding pose obtained in molecular docking is widely verified by molecular dynamics simulation (MDS) (Anusuya et al., 2015). The MD simulation utilized the Desmond package embedded in Maestro software. The ligands and the target were simulated in the water model TIP4P medium for two ns with ensemble NPT.
The stability of the docked complex was observed by measuring the root mean square deviation (RMSD) of each trajectory record for 4.8 ps (0.0048 ns) in the MD simulation concerning the initial position of the docked complex.
The RMSD of Cα and side chain atoms (part of the target) during the two ns period of MDS is shown in Fig. 4. The plot demonstrates that the RMSD of Cα and side-chain atoms for each ligand has a steep increase due to the initial binding to the substrate in the period of approximately 0.01 ns. Naturally, the target reaches stabilized confirmation after a few nanoseconds. Remdesivir takes 1.0 ns to reach a stabilized conformation and is restabilized again after 1.5 ns. Meanwhile, quercetin and hesperidin take one ns and 1.5 ns, respectively, to reach stabilized conformations. However, sulabiroin-A does not reach a stabilized conformation even after two ns.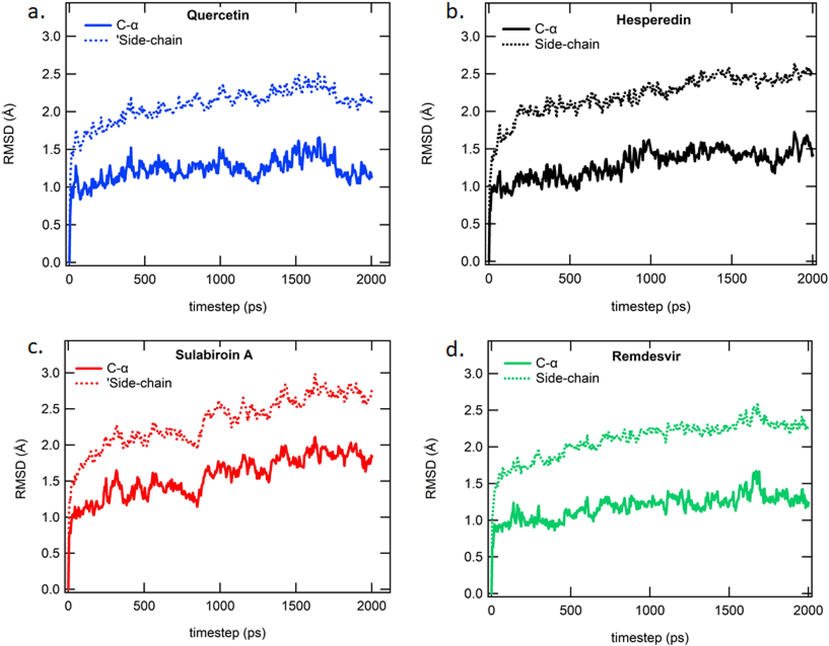
Protein RMSD for a 2 ns period of MD simulation studies for (a.) Quercetin (b) Hesperidin (c) Sulabiroin-A (d) Remdesivir.
The stability of the protein–ligand interaction can generally be observed from the RMSD path, as shown in Fig. 5. However, it is difficult to differentiate RMSD among the four protein–ligand interactions. Thus, we fitted those RMSD trajectories with the power function of Equation (2)
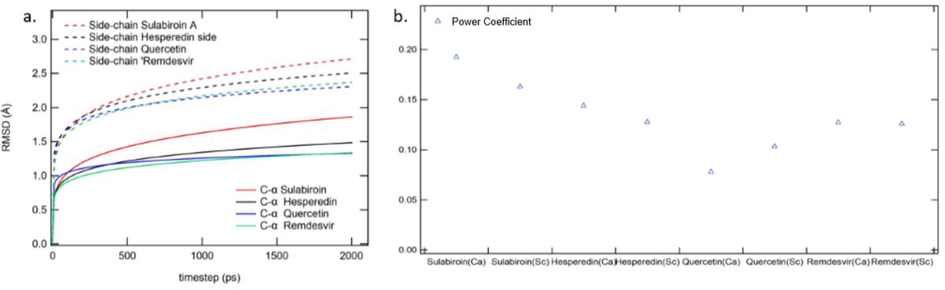
RMSD Analysis (a.) RMSD curve fitting (b) variable plotting.
Next, we present the results for RMSF (root mean square fluctuation). The RMSF for each ligand is shown in Fig. 6 (a), (b), (c), and (d). RMSF characterizes local changes along the protein chain. It is expected that the RMSF value in the terminal part is higher than that in the in-between part since these sites fluctuate more than other sites.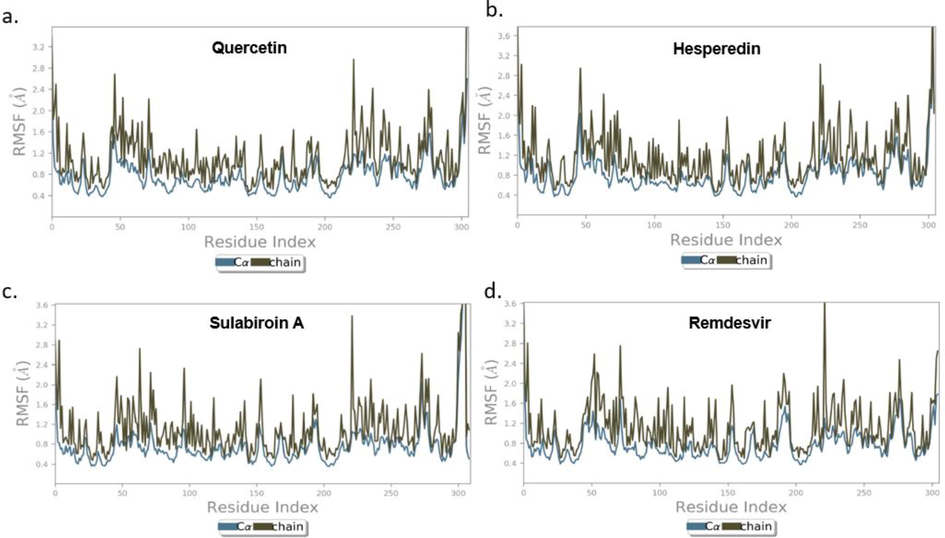
Protein RMSF for (a.) Quercetin (b) Hesperidin (c) Sulabiroin-A (d) Remdesivir.
On the other hand, the RMSF value for the in-between part is lower than that for the terminal part due to the rigid structure of the alpha-helix and beta-strand. However, some sites in the in-between region have an RMSF value higher than 2.5 Å, which indicates that these sites are not stable. The RMSF was observed to a minimum of 0.36 Å for Cα atoms and 0.47 Å for side-chain atoms, whereas maximum RMSFs of 2.2 Å and 3.6 Å were observed for Cα and side-chain atoms, respectively. In general, RMSF results showed that the ligand target is sufficiently stable, and the conformation has low flexibility.
To further verify that the protein–ligand complex is stable, we observed the RMSF protein at specific residues where interaction takes place. RMSF fluctuation is connected to the RMSD plot, in which more fluctuation in the RMSF plot generally yields a less stable RMSD plot. It is understood that RMSF gives local residual fluctuation, while RMSD gives global fluctuation. For example, the Sulabiroin-A-target RMSF has the most fluctuation in RMSF, thus giving the least stable RMSD plot. From the results, it can be observed that quercetin and hesperidin show the least fluctuation.
Next, we investigated protein–ligand contact simulation results to validate docking scores and MM-GBSA values. Figs. 7 and 8 demonstrate the number of contacts of each residue and protein–ligand contacts. Fig. 7 (a) shows that in the protein–quercetin contact, the dominant contribution to the interaction is given by the Glu166 residue, where H-bond and water bridge interactions form a 0.8 interaction fraction (80% of the total interaction), as shown in Fig. 8 (a). The other interactions are given by other residues that help the protein–ligand interaction, Thr25, His41, Ser46, Glu47, Gln189, and Asn142, where the combination of H-bonds and water bridges with interaction fractions of approximately 0.35 to 0.4.
Timeline representation of protein–ligand contact of (a) quercetin, (b) hesperidin, (c) sulabiroin-A, and (d) remdesivir.
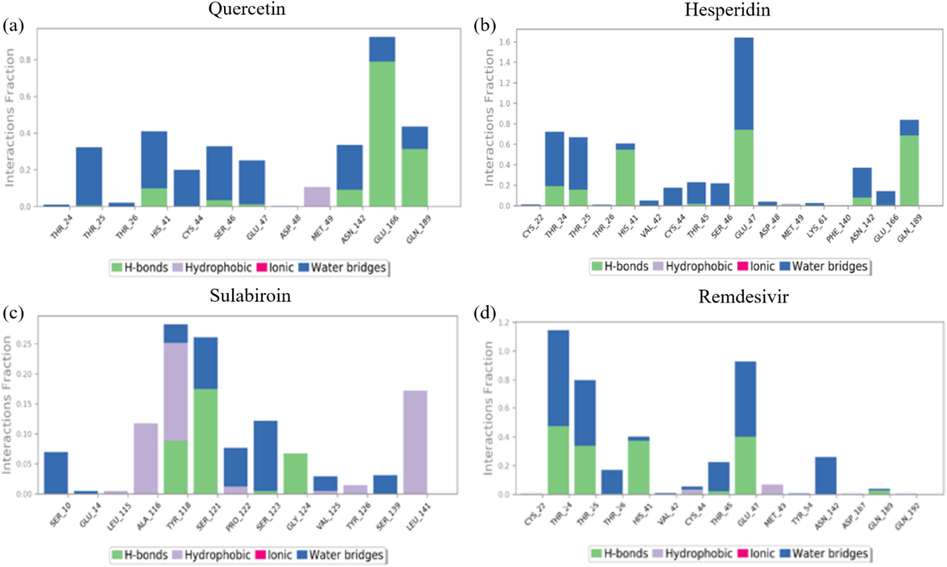
Protein-ligand contact of (a) quercetin, (b) hesperidin, (c) sulabiroin-A, and (d) remdesivir.
Fig. 7 (b) and 8 (b) show that the protein-hesperidin contact has the highest interaction overall, whether in several contacts and interaction fractions. The interaction fraction is approximately 1.6 for residue glu47, with the formation of hydrogen bonds and water bridges, whereas other residues of Thr24, Thr25, His41, and Gln189 gave interaction fraction values of approximately 0.65 to 0.8.
Fig. 7 (c) and 8 (c) show that the protein-sulabiroin-A contact has the least interaction compared to other ligands. The highest interaction fraction is only 0.25, given by Tyr118 and Ser121. All the other residues cannot attain beyond 0.2 of the interaction fraction. This result indicates that Sulabiroin-A has the least interaction with protein compared to other ligands.
Fig. 7 (d) and 8 (d) show the protein-remdesivir contact, and the interaction fraction is mainly given by Thr24, Thr25, and Glu47. These interactions are given by the formation of H-bonds and water bridges. The other interactions are given by another residue of His41, Thr45 with the formation of H-bonds and water bridges, and Asn142 with hydrogen bond formation. These interactions give interaction fractions of approximately 0.1 to 0.4.
Overall, the consistency of interaction between protein ligands has been found in hesperidin ligands, remdesivir ligands, and quercetin ligands in the order of degree of consistency. This means that the interaction between protein ligands occurs for most of the period in the simulation. Meanwhile, the least consistency was shown for Sulabiroin A-ligand. This means that the protein–ligand interactions do not occur continuously during the period in the simulation. It can be understood by the fact that in Sulabiroin-A, the interaction was driven by the hydrophobic part of the ligand.
From ligand-protein contact, Glu166, Glu47, and Thr24 dominantly interacted in quercetin, hesperidin, and remdesivir, and other less dominant residues were Asn142, His41, Gln189, Thr25, Thr45, and Ser46. To further verify that the protein–ligand complex interaction is stable, we evaluated the protein RMSF at specific residues where interaction takes place, as shown in Fig. 9. RMSF is a local fluctuation of each residue in the protein, and by these data, we can observe whether an individual residue of the protein target was stable during the simulation. The residues chosen were those with dominant fraction interactions of protein–ligand contacts. The RMSF of protein Ca of all complexes is shown in Fig. 9. Overall, the RMSF of protein–ligand contact shows a low value of RMSF (<1,5 Å), which indicates stable contact at the residue.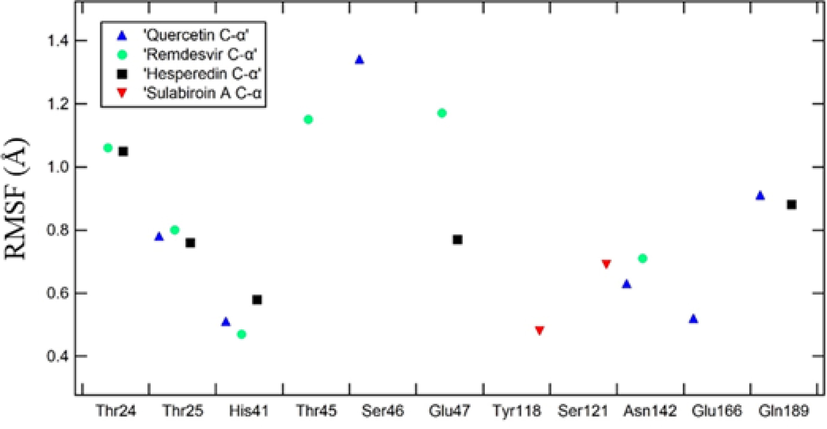
RMSF Ligand–specific residue interaction.
4 Conclusion
We successfully conducted molecular docking and molecular dynamics simulations on ligand-target CoV main protease ligands (quercetin, hesperidin, sulabiroin-A, and remdesvir). Based on molecular docking scores (docking score and MMGBSA), we found that all the ligands were excellent candidates as inhibitors. The docking scores order from high to low: Hesperidin, Remdesivir, Quercetin, and Sulabiroin-A and MMGBSA with similar order. These results place sulabiroin-A as a suitable inhibitor comparable with other ligands. The molecular dynamics simulation demonstrates that all the ligands can also serve as good candidates as inhibitors, although the fluctuation of sulabiroin-A remains quite high and has less protein–ligand interaction time than other ligands. However, the possibility of Sulabiroin-A being an alternative inhibitor is still widely open if the newer structure of SARS-CoV-2 main protease is used as an alternative target.
Based on the molecular dynamics simulation, sulabiroin-A has no interaction with essential amino acids in the binding site. Furthermore, sulabiroin-A may be a potential inhibitor of other SARS-CoV-2 proteins.
Disclosure of funding
There are no financial conflicts of interest to disclose. This study was funded by Publikasi Terindeks Internasional (PUTI) Q1 Grant Universitas Indonesia number NKB-1426/UN2.RST/HKP.05.00/2020.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Antimicrobial and immunomodulating activities of Hesperidin and ellagic acid against diarrheic Aeromonas hydrophila in a murine model. Life Sci.. 2013;93(20):714-722.
- [Google Scholar]
- Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300(5626):1763-1767.
- [Google Scholar]
- Quercetin derivatives as non-nucleoside inhibitors for dengue polymerase: molecular docking, molecular dynamics simulation, and binding free energy calculation. J. Biomol. Struct. Dyn.. 2017;35(13):2895-2909.
- [Google Scholar]
- Identification of dengue viral RNA-dependent RNA polymerase inhibitor using computational fragment-based approaches and molecular dynamics study. J. Biomol. Struct. Dyn.. 2015;34(7):1512-1532.
- [CrossRef] [Google Scholar]
- Review of the biological properties and toxicity of bee propolis (propolis) Food Chem. Toxicol.. 1998;36(4):347-363.
- [Google Scholar]
- A trial of lopinavir–ritonavir in adults hospitalized with severe Covid-19. N. Engl. J. Med.. 2020;382(19):1787-1799.
- [Google Scholar]
- Cavasotto C.N., ed. In silico drug discovery and design: theory, methods, challenges, and applications. CRC Press; 2015.
- Potential therapeutic agents for COVID-19 based on the analysis of protease and RNA polymerase docking. 2020.
- Breakthrough: Chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci. Trends 2020
- [Google Scholar]
- The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov.. 2015;10(5):449-461.
- [Google Scholar]
- MM/GBSA binding energy prediction on the PDBbind data set: Successes, failures, and directions for further improvement. J. Chem. Inf. Model.. 2013;53(1):201-209.
- [Google Scholar]
- Improved docking results via reranking of ensembles of ligand poses in multiple x-ray protein conformations with MM-GBSA. J. Chem. Inf. Model.. 2014;54(10):2697-2717.
- [Google Scholar]
- The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak–an update on the status. Mil. Med. Res.. 2020;7(1)
- [CrossRef] [Google Scholar]
- Subcritical water extraction of flavonol quercetin from onion skin. J. Food Eng.. 2011;102(4):327-333.
- [Google Scholar]
- Evaluating the potency of Sulawesi propolis compounds as ACE-2 inhibitors through molecular docking for COVID-19 drug discovery preliminary study. J. King Saud Univ. Sci. 2021;33(2):101297.
- [CrossRef] [Google Scholar]
- Antibacterial, antifungal and antiviral activity of propolis of different geographic origin. J. Ethnopharmacol.. 1999;64(3):235-240.
- [Google Scholar]
- Antiviral effects and possible mechanisms of action of constituents from Brazilian propolis and related compounds. J. Apic. Res.. 2020;59(4):413-425.
- [Google Scholar]
- Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov.. 2020;6(1):1-4.
- [Google Scholar]
- Advances in studies on the biological activities of Hesperidin and hesperetin. Chin. J. New Drugs. 2011;20(4):329-381.
- [Google Scholar]
- Clean recovery of antioxidant flavonoids from citrus peel: optimizing an aqueous ultrasound-assisted extraction method. Food Chem.. 2010;119(1):81-87.
- [Google Scholar]
- Ultrasound-assisted extraction of Hesperidin from Penggan (Citrus reticulata) peel. Ultrason. Sonochem.. 2008;15(3):227-232.
- [Google Scholar]
- Propolis Components and Biological Activities from Stingless Bees Collected on South Sulawesi, Indonesia. HAYATI J. Biosci.. 2020;27(1) 82 82
- [Google Scholar]
- Computational investigations of three main drugs and their comparison with synthesized compounds as potent inhibitors of SARS-CoV-2 main protease (Mpro): DFT, QSAR, molecular docking, and in silico toxicity analysis. Journal of King Saud Univ.-Sci.. 2021;33(2):101315.
- [CrossRef] [Google Scholar]
- Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 Protease against COVID-19. J. Biomol. Struct. Dyn. 2020:1-6.
- [Google Scholar]
- Molecular interaction analysis of Sulawesi propolis compounds with SARS-CoV-2 main protease as preliminary study for COVID-19 drug discovery. J. King Saud Univ. Sci.. 2021;33(1):101234.
- [CrossRef] [Google Scholar]
- Bridging molecular docking to molecular dynamics in exploring ligand-protein recognition process: an overview. Front. Pharmacol.. 2018;9:923.
- [Google Scholar]
- Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature. 2016;531(7594):381-385.
- [Google Scholar]
- Ultrasound-assisted extraction of rutin and Quercetin from Euonymus alatus (Thunb.) Sieb. Ultrason. Sonochem.. 2008;15(4):308-313.
- [Google Scholar]
- In vitro antioxidant and antimicrobial activities of the extract of Pericarpium Citri Reticulatae of a new Citrus cultivar and its main flavonoids. LWT-Food Sci. Technol.. 2008;41(4):597-603.
- [Google Scholar]
- Antiviral activity of hatay propolis against replication of herpes simplex virus type 1 and type 2. Med. Sci. Monitor: Int. Med. J. Exp. Clin. Res.. 2016;22:422.
- [Google Scholar]
- Antiviral activity of four types of bioflavonoid against dengue virus type-2. Virol. J.. 2011;8(1):1-11.
- [Google Scholar]
- Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368(6489):409-412.
- [Google Scholar]
- A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270-273.
- [Google Scholar]
- A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med.. 2020;382(8):727-733.
- [Google Scholar]




