

Mining novel natural reactive oxygen species (ROS) inhibitors by targeting Rho Kinase for prevention of secondary spinal cord injury: An in-silico trial using traditional Chinese medicinal compounds
⁎Corresponding author at: Department of Orthopaedic Surgery, Shuguang Hospital, Shanghai University of Traditional Chinese Medicine, No 528 of zhangheng road pudong new area, Shanghai 2010203, China. RonnyVaughanexo@yahoo.com (Shuqiang Wang)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Traditional Chinese Medicinal (TCM) compounds provide a plethora of natural chemiome for structure based novel drug discovery against unexplored targets of important diseases. One such disease is Secondary Spinal cord Injury (SSCI), a condition secondary to initial Spinal cord Injury (SCI) caused by a trauma. In SSCI oxidative stress and inflammation play a pivotal role in aggravating neural damage at the site of trauma. To look into it reactive oxygen species (ROS) inhibition is a good strategy. Our Study here focuses on finding novel ROS inhibitors from in-house TCM compound library using advanced structure based drug discovery methods. From Virtual screening, Molecular Docking, Molecular Dynamics Simulation and MM-PBSA calculations a single ROS inhibitor was proposed for targeting SSCI. Our study provides a platform for future structure based drug discoveries in the field of treating SCI by targeting SSCI pathways.
Keywords
Secondary spinal cord injury
Reactive oxygen species
Traditional Chinese medicine
Molecular docking

1 Introduction
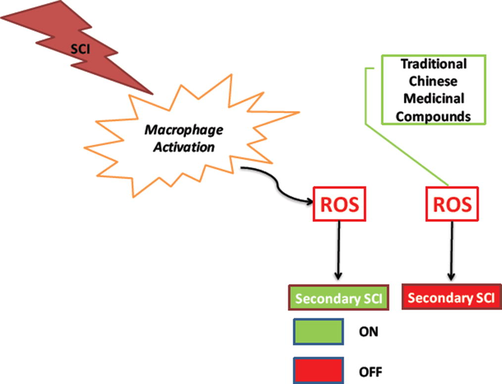
Spinal cord injury (SCI) is an injury triggered event that is associated with permanent neurologic deficit. The deficit instigated by SCI leads to medical comorbidity, not only effecting sensory and motor capabilities, but also having impact on the physiological and economical condition of the patient (McDonald and Sadowsky, 2002). Edwin Smith papyrus, an Egyptian physician in 1700 BCE was the first one to document SCI as an “ailment not to be treated. Since then SCI has been recorded as one of the devastating conditions where most of the cases are exanimate before any patient care is given. The surviving SCI cases remain morbid and are more prone to mortality,in USA alone 10–40 people in a million in a year are effected by SCI. The total number of cases estimated in USA as reported in 2016 are at staggering 282,000, to which every year 11,000 new cases are added. Billions of dollars in USA alone are spend on this disease, making it one of the economically devastating diseases. (White and Black, 2016).
SCI is categorized into primary spinal cord injury(pSCI)and secondary spinal cord injury(sSCI), pSCI is defined as the injury inflicted at the time of trauma and sSCI is defined by the injury causedby the body’s response to initial trauma (Cadotte and Fehlings, 2011). The consequences of the SCI are defined by the extent of secondary damage, which is initiated by a cascade of molecular cellular events triggered by pSCI. The pSCI triggers glutamatergic excitotoxicity, free radical damage, cytokine production and inflammation, all of them effecting the survival of neurons and glial cells, thus setting a base for onset of sSCI, leading to other patho-mechanisims that trigger neuropathic pain and autonomic dysfunction. Use of free-radical scavengers, anti-inflammatory drugs and anti-apoptotic drugs are suggested to be effective therapeutic strategies for the inhibition of sSCI (Zhou et al., 2014).
In the pathogenesis of sSCI the role of reactive oxygen species (ROS) molecules in oxidative damage to spinal cord lipids, known as lipid peroxidation (LP) is well established Hall et al., 2016). The hydrogen peroxide and peroxinitrites are non-radical ROS reported to play important roles in post pSCI onset, among them OH, NO2 and CO3 peroxinitrites are more prominent to initiate LP.The expression of peroxinitrites is regulated by RhoA GTPase (RhoA) / Rho-associated kinase (ROCK) pathway, other than this ROCK is also associated with cytoskeletal rearrangement and cell movement function in a cell. This RhoA/ROCK pathway is implicated in disorders like cardiovascular disease (Budzyn et al., 2006) and central nervous system diseases (Yamamoto et al., 2014). Spinal cord injury is one of the prominent CNS disease among others like multiple sclerosis, Alzheimer’s disease, glaucoma and stroke to be regulated by RhoA/ROCK pathway (Tokushige et al., 2011).
Computer aided drug designing (CADD) is a promising strategy in novel drug discovery for rare diseases. This powerful tool is an established standard for novel drug designing and discovery, novel leads have been reported as enzyme inhibitors as well as protein − protein interaction disruptors using this method (Amin et al., 2016). In this study we are trying to look into novel natural lead compound of traditional Chinese medicine (TCM) origin that will alter RhoA/ROCK pathway by inhibiting the ATP binding site of ROCK. The main aim will be to discover a compound that will alter ROS based sSSI. We have used TCM database of 672 compounds for multistep structure based CADD method, virtual screening technique, followed by docking and simulation was employed for identifying promising lead compound, the approach is very well used method for identifying novel leads.
2 Material and method
2.1 Protein and ligand preparation
For CADD the atomic coordinates for ROCK protein (PDB ID: 3V8S) was taken. The structure was checked for missing atoms and the complete structure was energy minimised using swiss PDB viewer (SPDBv) (Viewer et al., 2001). The RMSD (Root Mean Square Deviation) was monitored and using GROMOS96 43B1 force field (van Gunsteren et al., 1996). Six hundred and seventy two highly active TCM compounds from in-house database (Table 1) was used for targeting ATP binding site of ROCK protein.
| Ingredient_name |
|---|
| Chrysophanic acid |
| 4-hydroxybenzoic acid |
| Succinate |
| hexose |
| Aspartate |
| Glutamine |
| Hexadecenoic acid |
| Octadecenoic acid |
| Cardiolipin |
| glutamate |
| caffeic acid phenethyl ester |
| tretinoin |
| cytochalasin B |
| lovastatin |
| serine |
| gibberellic acid |
| Arabinose |
| benzoate |
| norethynodrel |
| 7-methoxycoumarin |
| 10-hydroxy-camptothecin |
| trans-p-Hydroxycinnamic acid |
| 2,6-Di-tert-butyl-4-methylphenol |
| ammonium glycyrrhizinate |
| palmatine chloride |
| acacetin |
| artemisin |
| atropine |
| avicularin |
| baicalin |
| belladonnine |
| biflorin |
| bilirubin |
| biochanin a |
| biotin |
| brucine |
| budlein a |
| butein |
| caffeine |
| camptothecin |
| catharanthine |
| cephalomannine |
| cholesterol |
| chrysin |
| colchicine |
| cortisone |
| coumestrol |
| cryptopine |
| cucurbitacin e |
| cucurbitacin i |
| curcumin |
| daidzein |
| digoxin |
| dubinidine |
| ellipticine |
| enhydrin |
| epicatechin |
| erysovine |
| erythraline |
| estrone |
| eucalyptin |
| formononetin |
| fructose |
| galangin |
| galanthamine |
| genipin |
| genistein |
| ginkgolide a |
| grandisin |
| guanidine |
| guanosine |
| harmaline |
| harmine |
| harringtonine |
| hesperetin |
| homoeriodictyol |
| homoharringtonine |
| honokiol |
| humulone |
| hyoscyamine |
| isoquercitrin |
| isovitexin |
| kaempferol |
| khellin |
| kinetin |
| lapachol |
| alpha-lapachone |
| beta-lapachone |
| licarin a |
| luteolin |
| maltose |
| mangiferin |
| morin |
| naringenin |
| nobiletin |
| orientin |
| perfamine |
| phytosphingosine |
| piceid |
| picrotin |
| picrotoxinin |
| piplartine |
| podophyllotoxin |
| porphyrin |
| precocene ii |
| pregnenolone |
| procyanidin b2 |
| tryptanthrin |
| gallic acid |
| epigallocatechin |
| salicylic acid |
| caffeic acid |
| ellagic acid |
| catechin |
| artemisinin |
| hyperoside |
| estradiol |
| pseudoephedrine |
| reynoutrin |
| asparagine |
| erythrinin |
| quinic acid |
| fluoxetine |
| nifedipine |
| methylprednisolone |
| galgravin |
| artesunate |
| artemether |
| melatonin |
| secoisolariciresinol |
| alternariol |
| velutin |
| vicenin-2 |
| mannitol |
| apigetrin |
| cholic acid |
| lithocholic acid |
| physostigmine |
| riboflavin |
| ginkgolide |
| quercitrin |
| reserpine |
| ribalinine |
| rutin |
| sanguinarine |
| sophocarpine |
| sphingomyelin |
| sucrose |
| swertisin |
| tanshinone i |
| tanshinone iia |
| taxol |
| tetrandrine |
| thebaine |
| theophylline |
| tiliroside |
| tremulacin |
| 7,3′,4′-trihydroxyflavone |
| 3,5,3′-triiodothyronine |
| triptolide |
| tropine |
| tryptanthrine |
| valine |
| veraguensin |
| vincristine |
| vitexin |
| yohimbine |
| 5-o-caffeoylquinic acid |
| ursodeoxycholic acid |
| taxifolin |
| sorbitol |
| icariin |
| rosmarinic acid |
| gallocatechin |
| (−)-epicatechin |
| (−)-noradrenaline |
| (+)-catechin |
| (+)-epicatechin |
| 1,16-hexadecanediol |
| 1,2-benzenediol |
| 11-deoxojervine |
| 15,16-dihydrotanshinone i |
| 17-hydroxycryptotanshinone |
| 1-hydroxyanthraquinone |
| 1-kestose |
| 1-ketoisocyptotanshinone |
| 2,5-dihydroxy benzoic acid |
| 2-acetamido-2-deoxy-d-glucose |
| 2′-deoxythymidine |
| 2-hydroxyanthraquinone |
| 2-hydroxybenzoic acid |
| 2-methoxycinnamic acid |
| 2-methyl-1,4-naphthoquinone |
| 3,3′,4′,5,5′,7-hexahydroxyflavone |
| 3,4-dihydroxybenzoic acid |
| 3,4-phenanthrenedione |
| 3,7,11,15-tetramethyl-2-hexadecen-1-ol |
| 3-hydroxycyptotanshinone |
| 3-hydroxy-glabrol |
| 3-hydroxykynurenine |
| 3-hydroxymethylenetanshinquinone |
| 3-hydroxytanshinone iib |
| 3-methylquercetin |
| 3′-o-acetylhamaudol |
| 3-phenyl-2-propen-1-ol |
| 4-coumaric acid |
| 4-hydroxy-3-methoxybenzaldehyde |
| 4-hydroxybenzoic acid |
| 4-hydroxybenzoylcholine |
| 4-hydroxyphenylacetic acid |
| 4-methylpyrazole |
| 5′-adenosine monophosphate |
| 5-methyluracil |
| 6,7-dihydroxycoumarin |
| 6-aminopurine |
| 6-methoxy-7-hydroxycoumarin |
| 8-geranyloxy psoralen |
| abscisic acid |
| acacetin |
| acaciin |
| acetylcholine |
| acrylic acid |
| adenine |
| adenosine |
| adonitol |
| aesculetin |
| aesculin |
| afzelin |
| agmatine |
| albiflorin |
| allantoin |
| allocryptopine |
| aloin |
| alpha-bisabolol |
| alpha-copaene |
| alpha-tocotrienol |
| alpha-tocotrienol |
| amber acid |
| amentoflavone |
| aminoacetic acid |
| aminopyrine |
| anabasine |
| aniline |
| anserine |
| anthranilic acid |
| apigenin |
| apigenin 7-o-beta-d-glucopyranoside |
| apigenin-7-o-glucoside |
| apigenin-7-o-neohesperidoside |
| apigetrin |
| arachidonic acid |
| asparagine |
| aspartic acid |
| aspidocarpine |
| astragalin |
| atenolol |
| atrazine |
| atropine |
| baicalin |
| behenic acid |
| benzaldehyde |
| benzoate |
| benzoic acid |
| benzophenone |
| berberine |
| bergapten |
| beta-alanine |
| beta-carotene-5,6-epoxide |
| betaine |
| beta-thujaplicin |
| beta-tocopherol |
| betonicine |
| bicuculline |
| biochanin a |
| biotin |
| boldine |
| brassicasterol |
| brucine |
| butanedioic acid |
| butyrate |
| butyric acid |
| cadaverine |
| caffeic acid |
| caffeine |
| caffetannic acid |
| callistephin |
| calycosin-7-o-beta-d-glucoside |
| camphene |
| canavanine |
| canthaxanthin |
| caproic acid |
| capsaicin |
| carnitine |
| carnosine |
| catechin |
| catechol |
| chalcone |
| chelidonine |
| chenodeoxycholic acid |
| chlorogenic acid |
| cholalic acid |
| choline |
| chrysanthemin |
| cinaroside |
| cinchonine |
| cinnamic alcohol |
| cirsimarin |
| cis-4-hydroxyproline |
| cis-9-octadecenoic acid |
| cis-aconitic acid |
| citrin |
| citrulline |
| cocaine |
| codeine |
| coniferaldehyde |
| coniferyl aldehyde |
| cordycepic acid |
| cosmosiin |
| coumarin |
| creatine |
| creatinine |
| crithmene |
| cyanidin-3-glucoside |
| cyanin |
| cyclamin |
| cyclopamine |
| cystathionine |
| daidzein |
| daidzin |
| danshenxinkun a |
| daphnetin |
| decanedioic acid |
| delphinidin |
| delphinidin-3-glucoside |
| delta-tocotrienol |
| deoxycholic acid |
| d-glucuronic acid |
| diazinon |
| dichlorvos |
| dihydrocapsacine |
| dihydrocapsaicin |
| dihydrochelerythrine |
| dihydromelilotoside |
| dihydroquercetin |
| dimethyl malate |
| diosmin |
| dopamine |
| dulcitol |
| emetine |
| emetine |
| enanthic acid |
| ephedrine |
| epicatechin |
| epiprogoitrin |
| eriodictyol |
| esculetin |
| esculin |
| eserine |
| estrone |
| ethanolamine |
| ferulic acid |
| fipronil |
| fisetin |
| flavanone |
| flavanone |
| flavin mononucleotide |
| fluoxetine |
| foliosidine |
| formononetin |
| fortunellin |
| galactitol |
| galactosamine |
| gamma-aminobutyric acid |
| gamma-linolenic acid |
| gamma-nonalactone |
| gamma-terpinene |
| gamma-tocotrienol |
| genistein |
| gentiobiose |
| gentisic acid |
| gibberellic acid |
| ginsenoside rb1 |
| ginsenoside rb2 |
| ginsenoside rc |
| ginsenoside rd |
| ginsenoside re |
| ginsenoside rg1 |
| glabrene |
| glabridin |
| glabrol |
| glucoerucin |
| gluconasturtiin |
| glucosamine |
| glucotropaeolin |
| glutamic acid |
| glutaric acid |
| glutathione |
| glycerin |
| glycerol |
| glycine |
| glycocholic acid |
| glycolic acid |
| glycyrrhetinic acid |
| glycyrrhizic acid |
| glycyrrhizin |
| glycyrrhizinate |
| glycyrrhizinic acid |
| gomisin e |
| gomisin f |
| gomisin g |
| gossypin |
| guanidine |
| guanosine |
| haplopine |
| harmaline |
| harman |
| harmane |
| heptadecane |
| heptanoic acid |
| hesperetin |
| hesperidin methyl chalcone |
| heteroauxin |
| hexanoic acid |
| hippuric acid |
| hirsutrin |
| hispaglabridin a |
| hispaglabridin b |
| homogentisic acid |
| homoorientin |
| homoserine |
| hymecromone |
| hyoscyamine |
| hyperin |
| hyperoside |
| hypoxanthine |
| hypoxanthine |
| icariin |
| indole |
| indole-3-acetonitrile |
| indole-3-carboxaldehyde |
| inosine |
| isoamylamine |
| isobetanin |
| isobutyric acid |
| isocitric acid |
| isoguvacine |
| isoliquiritin |
| isomaltose |
| isoorientin |
| isoquercetin |
| isorhamnetin |
| isorhamnetin-3-beta-d-galactopyranoside |
| isorhamnetin-3-o-glucoside |
| isorhamnetin-3-o-rutinoside |
| isosakuranetin |
| isovaleric acid |
| juniperic acid |
| kaempferide |
| kaempferitrin |
| kaempferol |
| kaempferol-3-o-glucoside |
| kaempferol-3-rhamnoside |
| kinetin |
| kynurenic acid |
| kynurenine |
| laudanosine |
| levodopa |
| l-homocysteine |
| l-homoserine |
| licochalcone b |
| lignoceric acid |
| linarin |
| linoleic acid |
| liquiritin |
| liquiritin apioside |
| lumichrome |
| lutein |
| luteolin |
| luteolin 7-beta-d-glucopyranoside |
| luteolin-4′-o-glucoside |
| luteolin-7-o-glucoside |
| luteoloside |
| lysine acid |
| malonic acid |
| malvin |
| m-coumaric acid |
| melatonin |
| meletin |
| mesaconic acid |
| methyl dihydrojasmonate |
| methyl octadecanoate |
| methyl salicylate |
| methyl stearate |
| methylprednisolone |
| metolachlor |
| miltirone |
| miscanthoside |
| monodydroxytanshinone i |
| morin |
| morphine |
| mucic acid |
| myo-inositol |
| myricetin |
| narcissin |
| naringenin |
| naringenin-7-o-glucoside |
| naringin |
| neoeriocitrin |
| neohesperidin |
| niacinamide |
| nicotiflorin |
| nicotinamide |
| nicotine |
| nicotinic acid |
| noradrenaline |
| norvaline |
| notoginsenoside r1 |
| o-aminophenol |
| o-coumaric acid |
| oenin |
| oleic acid |
| ononin |
| o-phenylenediol |
| orientin |
| orotic acid |
| oxalacetic acid |
| paeoniflorin |
| paeonin |
| palmatine |
| palmitoleic acid |
| pantothenic acid |
| p-coumaric acid |
| pelargonidin |
| pentanoate |
| pentanoic acid |
| peonidin |
| peoniflorin |
| peonin |
| petunidin |
| phenethylamine |
| phloretic acid |
| phloretin |
| phloridzin |
| phlorizin |
| phosphoenolpyruvate |
| p-hydroxybenzoic acid |
| pipecolic acid |
| piperazine |
| piperidine |
| p-methoxycinnamic acid |
| polyprenol |
| poncirin |
| procyanidin b1 |
| procyanidin b2 |
| procyanidin b3 |
| procyanidin b4 |
| procyanidin c1 |
| progoitrin |
| propranolol |
| prostaglandin e1 |
| protocatechuic acid |
| protopine |
| prunin |
| puerarin |
| putrescine |
| pyridoxine |
| pyrocatechol |
| pyroglutamic acid |
| quercetin |
| quercetin-3-arabinoside |
| quercetin-3-o-alpha-l-rhamnopyranoside |
| quercetin-3-o-rutinoside |
| quercetin-3-rhamnoside |
| quercetin-4′-glucoside |
| quercetrin |
| quercitin |
| querciturone |
| quinone |
| quisqualic acid |
| raffinose |
| raphanin |
| reserpine |
| resveratrol |
| retinol |
| reynoutrin |
| rhamnetin |
| rhoifolin |
| ribitol |
| riboflavin |
| ricinine |
| robinin |
| rosmarinic acid |
| rotenone |
| rutin |
| sabinene |
| salicylic acid |
| salsolinol |
| sanguinarine |
| saponarin |
| sarcosine |
| sarsasapogenin |
| sativin |
| schisantherin a |
| schisantherin b |
| scopoletin |
| scopolin |
| scoulerine |
| sebacic acid |
| sennoside a |
| serotonin |
| sinalbin |
| sinapaldehyde |
| sinapic acid |
| sinapine |
| sinapyl alcohol |
| sinigrin |
| sinomenine |
| sissotrin |
| smilagenin |
| solasodine |
| sophoricoside |
| sparteine |
| spermidine |
| spermine |
| spiraeoside |
| styrone |
| suberic acid |
| succinic acid |
| sulfanilic acid |
| synephrine |
| syringaldehyde |
| syringic acid |
| syringic aldehyde |
| syringin |
| tamarixetin |
| tanshindiol b |
| tanshinone i |
| tanshinone iia |
| tanshinone iib |
| tanshinone vi |
| taurine |
| taurocholic acid |
| thebaine |
| theobromine |
| theophylline |
| thymol |
| tiliroside |
| trans-2-hexenal |
| trans-aconitic acid |
| trans-cinnamaldehyde |
| trans-cinnamic acid |
| triacanthine |
| tribuloside |
| trifolirhizin |
| trigonelline |
| trijuganone b |
| tropine |
| tropinone |
| tryptamine |
| tyramine |
| uridylic acid |
| urocanic acid |
| veratramine |
| vincetoxicoside b |
| vitexin |
| xanthine |
| xanthohumol |
| xanthotoxin |
| xylitol |
| zearalenone |
| zeatin |
| zeaxanthin |
3 Virtual screening drug likeliness prediction
A total of 128 TCM compounds were shortlisted after virtual screening based on their binding energy (ΔG) calculations (Trott and Olson, 2010). The selected compounds were further limited by subjecting them to rules set by lipiski (Lipinski, 2004). The Lipinki Rule of five (RO5) parameters gave us five compounds for further analysis.
3.1 Molecular docking analysis
AutoDock 4.2 tool was employed for molecular docking study to achieve structure based drug againstcPLA2 protein (Morris et al., 2009). The tool calculates energy values by classification of energies as; internal energy, and torsional free energy.
ΔG represents the overall binding energy. ΔGvdw, ΔGhbond, ΔGelec represents Vander Waals, hydrogen bonding, and electrostatic energies respectively. ΔGtor represents translation and rotation and the term ΔGdesolv indicates the desolvation on binding and hydrophobic effect. Lamarckian genetic algorithm (GA) default parameters were used for calculating ΔG of each shortlisted compound. Grid box (60 × 60 × 60 A°) was build around the active site. Energy values generated and the binding mode with cPLA2 protein site was used to limit the compound to single molecule.
3.2 Molecular visualization:
The cPLA2-Lead4 complex was studied using visualization tools Pymol (DeLano, 2002) and Discovery Studio (Studio, 2013).
4 Result and discussion
Virtual Screening: A database of in-house highly active TCM compounds were used to inhibit the ROCK protein by targeting its ATP binding site (Fig. 1). ROCK is composed of a ATP binding and a catalytic domain as where phosphorylation takes place, shown in Fig. 2. The ATP binding domain was used to generate inhibitors against ROCK protein. This inhibition has a role in reducing sSCI induced tissue damage via reduction of LP and decrease in oxidative stress. To come up with a novel ROCK protein inhibitor virtual screening, drug-likeliness, docking and molecular dynamics simulation methods were used. Virtual screening helped us to limit the number of compounds from the 672 natural productsto 128, based on their binding energy (ΔG Kcal/mol).

Drug likeliness: To limit the focus on compounds that could be promising for further development, we checked each compound for drug-likeliness. Drug-likeliness of shortlisted compounds was defined by mutagenic and carcinogenic property and rule of five (RO5) set by Lipinski RO5 properties include number of hydrogen bond donor (HBD), number of hydrogen bond acceptor (HBA) molecular weight (MW) and octanol/water partition coefficient (logP), the permissible range is HBD ≤ 5, HBA ≤ 10, MW ≤ 500 Dalton and clog p ≤ 5. Table 2 shows drug-likeliness properties, five compounds were shortlisted on their drug-likeliness values. All the compounds are accommodating the values expected from typical drugs.
| Drug | Plant source | CID No. | CSID No. |
|---|---|---|---|
| Absinthin | Artemisia absinthium Linn | CID 442138 | |
| Aescin | Aesculus indica colebr. & Camb. (Hippocastanaceae) | CSID 23089563 | |
| Aesculin | Aesculus hippocastanum Linn | CID 5281417 | |
| Aglycone | Eryngium coeruleum Bieb. | CSID 16736194 | |
| Alantolactone | Inula racemosa HK. F. | CID 72724 | |
| Amaroswerin | Gentiana kurroo Royle | CID 45359883 | |
| Andromedotoxin (Acetyllandromedol) | Rhododendron campanulatum D. Don. | CSID 7827535 | |
| Apigenin | Meconopsis horridula | CID 5280443 | |
| Apigravin | Apium graveolens L. | CSID 30776837 | |
| Apiumoside (Apiin) | Apium graveolens L. | CSID 4444321 | |
| Arnidiol | Calendula officinalis Linn. | CID 470259 | |
| Artabsin | Artemisia absinthium L | CID 442146 | |
| Artemisinin | Artemisia drancunculus L. | CID 68827 | |
| Asarone | Acorus calamus Linn | CSID 552532 | |
| Ascaridol | Chenopodium ambrosioides L. | CID 10545 | |
| Astragalin | Aesculus indica colebr. & Camb. (Hippocastanaceae) | CID 5282102 | |
| Atisine | Aconitum heterophyllum Wallich ex Royle | CID 9548630 | |
| Atropine | Atropa acuminata | CID 174174 | |
| Avicularin | Polygonum aviculare Linn. | CID 5490064 | |
| Azulene | Achillea millefolium L. | CID 9231 | |
| Barrigenol A1 | Eryngium coeruleum Bieb. | CID 177603 | |
| Barringenol R1 | Eryngium coeruleum Bieb. | CID 44202129 | |
| β-Dihydrofucosterol (Azuprostat) | Euphorbia helioscopia Linn. | CID 457801 | |
| Berberine | Berberis aristata DC | CID 2353 | |
| Bergapten | Apium graveolens L. | CID 2355 | |
| Bergenin | Bergenia stracheyi Hook | CID 2356 | |
| Bikhaconitine | Aconitum violaceum Jacq. | CID 441713 | |
| Borneol | Prangos pabularia Lindl. | CID 64685 | |
| Camphene | Prangos pabularia Lindl. | CID 6616 | |
| Cannabinin | Cannabis sativus Linn. | CSID 8372337 | |
| Cannabinol | Cannabis sativus Linn. | CID 2543 | |
| Capillarin | Artemisia drancunculus L. | CSID 2340963 | |
| Carpesterol | Solanum xanthocarpum | CID 21155918 | |
| Carvacrol | Carum carvi Linn. | CID 10364 | |
| Carvone | Carum carvi Linn. | CSID 21106424 | |
| Celerin | Apium graveolens L. | CSID 137753 | |
| Choline | Dictamnus albus Linn. | CID 305 | |
| Chrysophanic Acid (Chrysophanol) | Rheum emodi Wall. | CID 10208 | |
| Citronellol | Mentha arvensis Linn. | CID 8842 | |
| Colchicine | Colchicum leteum Baker | CID 6167 | |
| Convolvulin (Convolvin) | Convolvulus arvensis L. | CSID 245689 | |
| Coriandrol | Coriandum sativum Linn. | CID 67179 | |
| Coumarin | Angelica glauca Edgew. | CID 323 | |
| Cryptopine | Fumaria indica L. | CID 72616 | |
| Cyanidin | Asparagus racemosus Willd. | CID 68247 | |
| Diosgenin | Dioscorea deltoidea Wall | CID 99474 | |
| Ecdysterone | Achyranthes aspera L. | CID 5459840 | |
| Emodin | Rheum emodii | CID 3220 | |
| Ephedrine | Ephedra gerardiana | CID 5032 | |
| Esculetin | Koelpinia linearis Pall. | CID 5281416 | |
| Etoposide | Podophyllum hexandrum Royle | CID 36462 | |
| Faradiel | Calendula officinalis Linn. | CID 122856 | |
| Filicin | Dryopteris filixmas L. | CID 197044 | |
| Fumaramine | Fumaria indica L. | CID 6450006 | |
| Gentianine | Gentiana kurroo Royle | CID 354616 | |
| Gentiopicrin | Gentiana kurroo Royle | CSID 32697064 | |
| Harmaline | Peganum harmala Linn. | CID 5280951 | |
| Harmalol | Peganum harmala Linn. | CID 5353656 | |
| Harmine | Peganum harmala Linn. | CID 5280953 | |
| Hetisine | Aconitum heterophyllum Wallich ex Royle | CSID 10226875 | |
| Hetisinone | Aconitum heterophyllum Wallich ex Royle | CSID 10226887 | |
| Hexacosane | Anagallis arvensis L. | CSID 11901 | |
| Hyoscine | Datura stramonium Linn | CID 3000322 | |
| Hyoscyamine | Datura stramonium Linn | CID 64692 | |
| Hyperoside | Asparagus racemosus Willd. | CID 5281643 | |
| Imperialine (Kashmirine) | Fritillaria imperialis Linn. | CID 442977 | |
| Indaconitine | Aconitum violaceum Jacq. | CID 441740 | |
| Inokosterone | Achyranthes aspera L. | CID 441828 | |
| Intybin | Cichorium intybus L. | CID 174863 | |
| Irigenin | Iris kashmiriana | CID 5464170 | |
| Isoalantolactone | Inula racemosa HK. F. | CID 73285 | |
| Isoatisine | Aconitum heterophyllum Wallich ex Royle | CID 245006 | |
| Isoimperatorin | Anthriscus nemorosa Spreng | CID 68081 | |
| Isopimpinellin | Apium graveolens L. | CID 68079 | |
| Kaempferol | Anagallis arvensis L. | CID 5280863 | |
| Lactucin | Cichorium intybus L. | CID 3756497 | |
| Lactucopicrin | Lactuca serriola Linn. | CSID 2723771 | |
| Laureline | Skimmia laureola Hk. f. | CID 821373 | |
| Lignans | Daphne oleoides | CID 9917980 | |
| Luteolin | Meconopsis horridula | CID 5280445 | |
| Malvalic Acid | Althaea officinalis L. | CID 10416 | |
| Marrubin | Marrubium vulgare L. | CSID 66118 | |
| Maslinic Acid | Epilobium angustifolium Linn. | CID 73659 | |
| Mezerein | Daphne oleoides | CID 9549167 | |
| Myrcene | Prangos pabularia Lindl. | CID 31253 | |
| Nepetalactone | Nepeta cataria | CID 161367 | |
| Obaculactone (Dictamnolactone) | Dictamnus albus Linn. | CID 65071 | |
| Obtusilobin (Obtusifolin) | Anemone obtusiloba D. Don | CID 3083575 | |
| Oleanolic Acid | Epilobium angustifolium Linn. | CID 10494 | |
| Osthenol | Apium graveolens L. | CID 5320318 | |
| p-Cymene | Thymus serpyllum Linn. | CID 7463 | |
| Peganine | Peganum harmala Linn. | CID 72610 | |
| Pinoresinol | Daphne oleoides | CID 234817 | |
| Podophyllotoxin | Podophyllum hexandrum Royle | CID 10607 | |
| Prangolarin | Anthriscus nemorosa Spreng | CID 17536 | |
| Protopine | Argemone mexicana L. | CID 4970 | |
| Quercetin | Aesculus indica colebr. & Camb. (Hippocastanaceae) | CID 5280343 | |
| Rutin | Aesculus indica colebr. & Camb. (Hippocastanaceae) | CID 5280805 | |
| Sabinen | Nepeta cataria | CID 18818 | |
| Safranal | Crocus sativus L. | CID 61041 | |
| Sanguinarine | Fumaria indica L. | CID 5154 | |
| Santonin | Artemisia maritima Linn | CID 221071 | |
| scopoletin | Artemisia drancunculus L. | CID 5280460 | |
| Sesamin | Daphne oleoides | CID 72307 | |
| Seselin | Apium graveolens L. | CID 68229 | |
| Sesquiterpene | Acorus calamus L. | CSID 19953446 | |
| shikonin | Arnebia guttata Bunge | CID 479503 | |
| Sitosterol | Adonis aestivalis L. | CID 222284 | |
| Spathulenol | Nepeta cataria | CID 522266 | |
| Stigmasterol | Asparagus racemosus Willd. | CID 5280794 | |
| Taraxacin | Taraxacum officinale | CID 5241825 | |
| Taraxasterol | Taraxacum officinale | CID 5270604 | |
| Tectoreginin | Iris kashmiriana | CID 5281811 | |
| Trigonelline | Achillea millefolium L. | CID 5570 | |
| Tropane | Atropa acuminata | CID 637986 | |
| Umbelliferone | Skimmia laureola Hk. f. | CID 5281426 | |
| Ursolic Acid | Epilobium angustifolium Linn. | CID 64945 | |
| Valepotriate | Valeriana jatamansi Jones | CID 442436 | |
| Xylopinine (Govanine) | Corydalis govaniana | CID 226520 | |
| 1-Hentriacontanol | Aesculus indica colebr. & Camb. (Hippocastanaceae) | CSID 61640 | |
| 1,4-Cineole (Natural) | Artemisia maritima L. | CID 10106 | |
| 7-Methoxycoumarin (herniarin) | Artemisia drancunculus L. | CID 10748 | |
| 16-Hentriacontanone (palmitone) | Aesculus indica colebr. & Camb. (Hippocastanaceae) | CSID 85480 |
Molecular Docking: The five final shortlisted natural compounds from IBS database were docked using AutoDock 4.2 tool into the optimized binding site of ROCK protein (Fig. 3). In Table 3 we have shown the results generated. Three of the Five natural compounds were found to form hydrogen bond with ROCK protein (Table 4). AutoDock tool was used for molecular docking simulations, the top binding pose based on ΔG were taken for further analysis. Each binding pose was studied using discovery studio, the default parameters were used to calculate all the possible interactions. The interactions studied are van der waals, conventional hydrogen bond, carbon hydrogen bond, pi-cation, pi-donor hydrogen bond, alkyl and pi- alkyl interaction. The lead2-ROCK complex has binding energy of −7.34 Kcal/mol andis forming two conventional hydrogen bonds with TYR96 and HIS62 of cPLA2′s C2 domain. The binding pocket of lead2 (1-cyclohexyl-5-(4-methoxybenzyl)-5-(((1R)-8-oxo-5,6-dihydro-1H-1,5-methanopyrido[1,2-a][1,5]diazocin-3(2H,4H,8H)-yl) methyl)pyrimidine-2,4,6(1H,3H,5H)-trione) comprises following amino acids TYR96, VAL97, ASP40, LYS32, THR41, PRO42, ASP43, HIS62, ASN64, ASN65, ASP93, ALA94, and ASN95. The O22 atomic site of lead2 shows hydrogen bond interaction with TYR96 and HIS62, with a distance between the lead and ROCK of 1.88 Å and 1.67 Å respectively. Lead 4 (3-(furan-2-yl)-N-(furan-2-ylmethyl)-3-(p-tolyl)propan-1-amine) shows three conventional hydrogen bond interactions with the ATP binding domain of ROCK. Three atoms of lead4, N15, O2 andO5 are forming the bond with TYR96, HIS62 and ASN95 with a bond length of 1.82 Å, 2.14 Å and 1.97 Å respectively. The binding pocket of lead 4 comprises of nine amino acids: ASP40, THR41, ASN65, ASP43, ASN64, HIS62, ASN95, TYR96, and VAL97. Lead4 is having ΔG of −10.09 Kcal/mol, the best reported among the top ten compounds. The third compound showing interaction is Lead6 ((12bS)-7-(2-ethoxy-3-methoxyphenyl)-2-(3-isopropoxypropyl)-12b-methyl-2,3,6,7-tetrahydropyrazino [1′,2′:1,2] pyrido[3,4-b]indole-1,4(12H,12bH)-dione),the binding pocket of the lead 6 molecule with lipid binding C2 domain of CPLA2 comprises of following amino acids viz. TYR96, ALA94, ASN95, LEU39, LYS32, ASP40, ASP43, ASN65, ASN64 and HIS62. Out of them lead6 forms hydrogen bond with TYR96 and HIS62, the interaction of our interest here is formed by lead6 O16 and O23 position with TYR96 and HIS62 at bond length 1.31 Å and 2.01 Å respectively. Based on ΔG and the number of interactions lead 7 (1-((S)-2-amino-4-methylpentanoyl)-N-((S)-1-((4-fluorobenzyl)amino)-3-methyl-1-oxobutan-2-yl)piperidine-4-carboxamide hydrochloride) is the least ranked among the top ten natural compounds inhibiting ATP binding domain of ROCK. Its binding pocket comprises of eleven amino acids; ASN64, ASP43, ASN65, LYS32, MET38, GLY36, LEU39, ASP37, THR41, ASP93, ASN95, TYR96, and ALA94. Lead7 shows ΔG of −6.07 Kcal/mol and has single hydrogen bond interaction between lead4′s O13 position and CPLA2′s TYR96 with bond length of 1.52.

| Compound | Gibbs Free Energy (kcal/mol) | Drug-likeness | Absorbtion Distribution Metabolisim Excretion | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutagenicity | Carcinogenicity | HBA | HBD | TPSA | MW | BBB | Caco2 | HIA | MDCK | PPB | ||
| Carpesterol | −13.3787 | yes | No | |||||||||
| Peganine | −11.0601 | Yes | No | |||||||||
| Cyanidin | −10.4529 | Yes | No | |||||||||
| Isoimperatorin | −10.3158 | Yes | No | |||||||||
| Capillarin | −10.2923 | Yes | Yes | |||||||||
| Atropine | −10.195 | No | Yes | |||||||||
| Taraxacin | −10.0508 | Yes | Yes | |||||||||
| Aescin | −10.0481 | |||||||||||
| Irigenin | −10.0414 | No | No | Q | Q | Q | Q | 0.043263 | 9.21056 | 86.80184 | 1.77078 | 79.69777 |
| Hyoscyamine | −10.0339 | Yes | No | |||||||||
| Osthenol | −9.58138 | Yes | No | |||||||||
| Sabinen | −9.50068 | Yes | No | |||||||||
| p-cymene | −9.42523 | Yes | Yes | |||||||||
| Sanguinarine | −9.35777 | Yes | Yes | |||||||||
| Coumarin | −9.33665 | Yes | Yes | |||||||||
| Safranal | −9.29266 | No | No | Q | Q | Q | Q | 1.06267 | 23.0033 | 100 | 249.139 | 15.17273 |
| Cannabinol | −9.18551 | No | Yes | |||||||||
| Myrcene | −9.10893 | Yes | No | |||||||||
| Ephedrine | −9.01662 | Yes | No | |||||||||
| Carvone | −8.96438 | Yes | No | |||||||||
| Intybin | −8.92803 | No | Yes | |||||||||
| Azulene | −8.92402 | Yes | Yes | |||||||||
| Carvacrol | −8.89384 | Yes | No | |||||||||
| Faradiol | −8.89069 | No | Yes | |||||||||
| Bergenin | −8.86525 | Yes | No | |||||||||
| Hyperoside | −8.78649 | No | No | V | V | Q | Q | |||||
| Avicularin | −8.67923 | Yes | No | |||||||||
| Taraxasterol | −8.49042 | Yes | No | |||||||||
| Emodin | −8.43599 | No | No | Q | Q | Q | Q | 0.668094 | 20.2745 | 90.42972 | 44.9367 | 100 |
| Maslinic Acid | −8.40496 | No | Yes | |||||||||
| Kaempferol | −8.36988 | Yes | No | |||||||||
| Asarone | −8.34362 | Yes | Yes | |||||||||
| Ecdysterone | −8.33694 | No | Yes | |||||||||
| Apigenin | −8.32192 | Yes | No | |||||||||
| Obaculactone | −8.29701 | Yes | No | |||||||||
| Umbelliferone | −8.281 | Yes | Yes | |||||||||
| Amaroswerin | −8.23858 | No | Yes | |||||||||
| Tectorigenin | −8.23005 | No | No | Q | Q | Q | Q | 0.126227 | 5.59415 | 88.18405 | 16.8164 | 87.63624 |
| Oleanolic Acid | −8.21107 | No | Yes | |||||||||
| Celerin | −8.15443 | Yes | No | |||||||||
| Barringenol A1 | −8.02007 | No | Yes | |||||||||
| Obtusifolin | −8.00917 | Yes | No | |||||||||
| Esculetin | −7.94481 | |||||||||||
| 7-Methoxycoumarin | −7.94033 | |||||||||||
| Arnidiol | −7.92807 | |||||||||||
| Chrysophanic Acid | −7.90567 | |||||||||||
| Astragalin | −7.90475 | |||||||||||
| Fumaramine | −7.8825 | |||||||||||
| Ursolic Acid | −7.83721 | |||||||||||
| Sitosterol | −7.78748 | |||||||||||
| Marrubin | −7.76988 | |||||||||||
| Aesculin | −7.76358 | |||||||||||
| Shikonin | −7.70705 | |||||||||||
| Hetisine | −7.62995 | |||||||||||
| Seselin | −7.57116 | |||||||||||
| Ascaridol | −7.54342 | |||||||||||
| Hyoscine | −7.5294 | |||||||||||
| Malvalic Acid | −7.48294 | |||||||||||
| Quercetin | −7.4617 | |||||||||||
| Inokosterone | −7.41245 | |||||||||||
| Luteolin | −7.39778 | |||||||||||
| Imperialine | −7.38899 | |||||||||||
| Santonin | −7.25135 | |||||||||||
| Camphene | −7.22408 | |||||||||||
| Stigmasterol | −7.2086 | |||||||||||
| Diosgenin | −7.20553 | |||||||||||
| Sesquiterpene | −7.19878 | |||||||||||
| Isopimpinellin | −7.10912 | |||||||||||
| Absinthin | −7.08183 | |||||||||||
| Filicin | −7.06472 | |||||||||||
| Colchicine | −7.05574 | |||||||||||
| Isoatisine | −6.99133 | |||||||||||
| Laureline | −6.98638 | |||||||||||
| Lactucopicrin | −6.96841 | |||||||||||
| ß-Dihydrofucosterol | −6.95752 | |||||||||||
| Scopoletin | −6.88906 | |||||||||||
| Andromedotoxin | −6.86834 | |||||||||||
| Gentiopicrin | −6.84314 | |||||||||||
| Hetisinone | −6.83401 | |||||||||||
| Harmalol | −6.82384 | |||||||||||
| Podophyllotoxin | −6.82185 | |||||||||||
| Harmaline | −6.81269 | |||||||||||
| Govanine | −6.78949 | |||||||||||
| Bergapten | −6.75754 | |||||||||||
| Lactucin | −6.75577 | |||||||||||
| Sesamin | −6.74654 | |||||||||||
| Harmine | −6.72931 | |||||||||||
| Protopine | −6.71054 | |||||||||||
| Apiin | −6.68698 | |||||||||||
| Artabsin | −6.64119 | |||||||||||
| Berberine | −6.62831 | |||||||||||
| Valepotriate | −6.59665 | |||||||||||
| Barringenol A1 | −6.59396 | |||||||||||
| Apigravin | −6.52009 | |||||||||||
| Artemisinin | −6.51894 | |||||||||||
| Spathulenol | −6.4596 | |||||||||||
| Alantolactone | −6.43428 | |||||||||||
| Isoalantolactone | −6.42729 | |||||||||||
| Convolvin | −6.33586 | |||||||||||
| Rutin | −6.23839 | |||||||||||
| Pinoresinol | −6.23388 | |||||||||||
| Citronellol | −6.21042 | |||||||||||
| Nepetalactone | −6.19349 | |||||||||||
| Atisine | −6.18617 | |||||||||||
| 1,4-Cineole | −6.02481 | |||||||||||
| Tropane | −5.85134 | |||||||||||
| Borneol | −5.81539 | |||||||||||
| Prangolarin | −5.73041 | |||||||||||
| Cryptopine | −5.59433 | |||||||||||
| Coriandrol | −5.59364 | |||||||||||
| Trigonelline | −5.55835 | |||||||||||
| 16-Hentriacontanone | −5.45843 | |||||||||||
| Lignans | −5.4099 | |||||||||||
| Hexacosane | −5.35611 | |||||||||||
| Cannabinin | −5.15507 | |||||||||||
| Mezerein | −4.89756 | |||||||||||
| Choline | −4.69109 | |||||||||||
| Etoposide | −4.47279 | |||||||||||
| 1-Hentriacontanol | −1.20476 | |||||||||||
| Aglycone | 24.0676 | |||||||||||
Q: Qualified; V: Violated.
| NAME | Chem ID | ΔG Kcal/mol |
Ligand binding pocket | H-bonds |
|---|---|---|---|---|
| Irigenin | 5,464,170 | −10.04 | GLY42, ILE43,PHE47,GLU45,HIS44 | IRIGENIN:H31 -:GLU45:O(2.082 Å). GLU45:H – : IRIGENIN:O3(1.81 Å). GLU45:H – : IRIGENIN:O4(2.29 Å). HIS44:HD1 – : IRIGENIN:O3(2.19 Å). HIS44:HD1 – IRIGENIN:O4(1.89 Å). IRIGENIN:H33 – ILE43:O (2.19 Å). ILE43:H – : IRIGENIN:O6(2.43 Å). |
| Safranal | 61,041 | −9.29 | HIS44, TYR68, LEU46, GLY61, TYR36, LEU62, LEU59 | TYR36:HH – :SAFRANAL:O1(1.99 Å). |
| Emodin | 3220 | −8.43 | ASP41, GLY42, ILE43, GLU45, HIS44 | EMODIN:H30 – GLU45:OE1 (2.03 Å). ILE43:H – : EMODIN:O2. (2.37 Å). EMODIN:H28 – A:ASP41:O(1.84 Å). |
| Tectorigenin | 5,281,811 | −8.23 | GLU45,ILE43,GLY42,ASP41,HIS44 | Tect:H34 – GLU45:OE1(1.91 Å). Tect:H29 – :ILE43:O(2.12 Å). ILE43:H – :TECT:O3(2.05 Å). |
Novel ROCK Inhibitor: The top natural compound inhibiting ATP binding domain of ROCK. The compound has the best binding energy and forms maximum number of hydrogen bonds, thus jamming the important ATP binding site in ROCK protein. To look into Lead 4 drug-ability, its Absorption, Distribution, Metabolism and Excretion (ADME) properties were calculated using in-silico ADME/Tox server (https://preadmet.bmdrc.kr/). In this calculation features like Caco-2 cell permeability (Caco-2p), MDCK cell permeability (MDCKp), Human intestinal absorption, Plasma Protein Binding and Blood Brain Barrier values of Lead 4 were calculated. The results generated are shown in Table 5. For drug absorption Caco-2 cell model and MDCK cell model were used and the value ranges in permissible range, the human intestinal absorbance (HIA) of 92.59% shows that Lead 4 can be well absorbed and can reach the target site easily. The plasma protein binding of lead 4 is 17.71% and shows its availability to reach the target protein is high. Evaluation of cell cytotoxicity revealed the IC50 for lead4 at 134.2 ± 6.8 μg/ml.
| Summary | Values |
|---|---|
| Van der Waal energy | −160.104 ± 23.737 kJ/mol |
| Electrostatic energy | −8.257 ± 8.986 kJ/mol |
| Polar solvation energy | 39.374 ± 14.616 kJ/mol |
| SAV energy | −92.616 ± 17.234 kJ/mol |
| Binding energy | −221.602 ± 35.657 kJ/mol |
Conflict of interest
The authors declared no conflict in this manuscript and publications.
References
- Irigenin, a novel lead from Western Himalayan chemiome inhibits Fibronectin-Extra Domain A induced metastasis in Lung cancer cells. Sci. Rep. 2016:6.
- [Google Scholar]
- Targeting Rho and Rho-kinase in the treatment of cardiovascular disease. Trends Pharmacol. Sci.. 2006;27(2):97-104.
- [Google Scholar]
- Spinal cord injury: a systematic review of current treatment options. Clin. Orthopaedics Related Res.®. 2011;469(3):732-741.
- [Google Scholar]
- DeLano, W.L., 2002. The PyMOL molecular graphics system.
- Lipid peroxidation in brain or spinal cord mitochondria after injury. J. Bioenerg. Biomembr.. 2016;48(2):169-174.
- [Google Scholar]
- Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov. Today: Technol.. 2004;1(4):337-341.
- [Google Scholar]
- AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem.. 2009;30(16):2785-2791.
- [Google Scholar]
- Studio, D., 2013. Accelrys Inc. San Diego, CA, USA.
- Effects of Y-39983, a selective Rho-associated protein kinase inhibitor, on blood flow in optic nerve head in rabbits and axonal regeneration of retinal ganglion cells in rats. Curr. Eye Res.. 2011;36(10):964-970.
- [Google Scholar]
- van Gunsteren, W.F. et al., 1996. Biomolecular simulation: the {GROMOS96} manual and user guide.
- Briefings Bioinf.. 2001;2(2):195-197.
- AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem.. 2010;31(2):455-461.
- [Google Scholar]
- White, N.-H., Black, N.-H., 2016. Spinal cord injury (SCI) facts and figures at a glance.
- The Novel Rho Kinase (ROCK) Inhibitor K-115: a new candidate drug for neuroprotective treatment in glaucomanovel rho kinase inhibitor. Invest. Ophthalmol. Vis. Sci.. 2014;55(11):7126-7136.
- [Google Scholar]
- Function of microglia and macrophages in secondary damage after spinal cord injury. Neural Regener. Res.. 2014;9(20):1787.
- [Google Scholar]




