

Translate this page into:
Machine learning approaches to genome-wide association studies
⁎Corresponding author at: Department of Computer and Information Sciences, Covenant University, Ota, Nigeria. vcosamor@gmail.com (Victor Chukwudi Osamor) victor.osamor@covenantuniversity.edu.ng (Victor Chukwudi Osamor)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Genome-wide Association Studies (GWAS) are conducted to identify single nucleotide polymorphisms (variants) associated with a phenotype within a specific population. These variants associated with diseases have a complex molecular aetiology with which they cause the disease phenotype. The genotyping data generated from subjects of study is of high dimensionality, which is a challenge. The problem is that the dataset has a large number of features and a relatively smaller sample size. However, statistical testing is the standard approach being applied to identify these variants that influence the phenotype of interest. The wide applications and abilities of Machine Learning (ML) algorithms promise to understand the effects of these variants better. The aim of this work is to discuss the applications and future trends of ML algorithms in GWAS towards understanding the effects of population genetic variant. It was discovered that algorithms such as classification, regression, ensemble, and neural networks have been applied to GWAS for which this work has further discussed comprehensively including their application areas. The ML algorithms have been applied to the identification of significant single nucleotide polymorphisms (SNP), disease risk assessment & prediction, detection of epistatic non-linear interaction, and integrated with other omics sets. This comprehensive review has highlighted these areas of application and sheds light on the promise of innovating machine learning algorithms into the computational and statistical pipeline of genome-wide association studies. This will be beneficial for better understanding of how variants are affected by disease biology and how the same variants can influence risk by developing a particular phenotype for favourable natural selection.
Keywords
Genome-wide association studies
Machine learning
Statistical learning
Single neuclotide polymorphism
Risk prediction
Epistasis

1 Introduction
Machine learning (ML) has been known to have to be useful for the analysis of whole genome data sets (including whole exome sequencing datasets), sequence annotation, epigenetic, proteomic and metabolomic data. As previously stated, they may also be referred to as statistical learning methods. As in genome-wide association studies (GWAS), we attempt to predict a phenotype from a genotype, such as the same in prediction problems of statistical learning. The primary purpose of machine learning algorithms is to define a function which predicts an unknown phenotype based on a genotype observation based on sample data (Mieth et al., 2016). A major reason for the adoption of machine learning algorithms is that they are well suited for developing predictive models when the number of features is larger than the number of samples. This is an important consideration when the GWAS SNP array datasets are generated for regular statistical testing in GWAS which detects only a few variants that can pass the stringent genome-wide significant level (e.g. p <10−8). In contrast, ML algorithms are focused on maximizing the prediction accuracy at the level of individual subjects (Okser et al., 2013). ML approaches are a worthy alternative in that they can perform significant attribute selection. They can also identify complex interactions between attributes, such as random forests, gradient boosting, and neural networks. A standard characteristic of GWAS results is that the number of attributes (p) greatly outnumber the number of sample points (n). This is usually described as the curse of dimensionality or the large p and small n problem. Ideally, it is a problem for classical multivariate regression. Another significant distinction between conventional factual strategies and ML techniques is that ML techniques don’t expect assumptions to be made about the hereditary components of an attribute being referred to. Attributes, for example, additivity of effects, the number and size of interactions and scope of interactions (Grinberg et al., 2020). Compared to univariate analysis of GWAS, ML models have provided a better means of learning multi-locus genetic variants as well as their interactions that predict complex traits (Okser et al., 2014). Furthermore, even though common regression methods have been used to characterize gene-gene interaction, they are very useful in searching for SNP combinations in high dimensional GWAS datasets (Szymczak et al., 2009). Machine Learning methods have been able to detect nearly all the previously identified variants by GWAS. This includes the best predictors and additional predictors with lower effects (Romagnoni et al., 2019). ML approaches that can obtain predictive models via training from prior genetic data have been recently applied to find significant SNPs for GWAS (Behravan et al., 2018; Mieth et al., 2016; Okser et al., 2014; Szymczak et al., 2009). In GWAS, genotypes of up to 1,000,000 SNPs are resolved in a few thousand subjects, prompting the little n, huge p issue (a lot more factors (SNPs) than tests). Secondly, when an enormous number of SNPs are genotyped on a genome-wide scale, linkage disequilibrium between SNPs should be considered. Therefore, standard multi-variable measurable methodologies like multiple linear regression or logistic regression are not appropriate for genome-wide data (Szymczak et al., 2009). Machine learning models have been shown to be able to capture the multi-locus SNPs interaction better than the regular univariate association studies. This has caused an increase in interest in this direction. Standard statistical genetics approaches have begun to be supplemented with, or even supplanted by, ML algorithms since they regularly make negligible suppositions about the fundamental disease mechanism, which is commonly obscure (Wang et al., 2013). ML algorithms apply multivariate, non-parametric approaches which identify patterns from data that is not normally distributed that are also strongly correlated (Ho et al., 2019). Okser et al. (2014) contended clinical utilizations of ML models in genetic disease risk prediction rely greatly on two elements, effective model regularization and thorough model validation. Maciukiewicz et al. (2018) studied major depressive disorder. Wherein they evaluated the possibility of using GWAS data for prediction of duloxetine outcomes with ML models. SVM and Classification trees models were used to predict response and remission while statistical learning approach, LASSO regression was used for feature selection in this experiment. It is imperative to review the machine learning approaches that have been applied in GWAS. The intended contribution of this work is to systematically map the specific application areas where machine learning algorithms have been applied in genome-wide association studies. This work further investigates the challenges and future trends of ML in GWAS in studing population variants and development of characteristics for natural selection. This is to enable the researcher to have a stronghold on the current literature and define the proper directions to develop novel computational and statistical approaches.
In order to treat GWAS as a machine learning problem as in Table 1, it is necessary to view the terms of GWAS as their machine learning counterparts. A genotype in GWAS terminology can be denoted as a feature in ML problems. In classification problems, there is usually a decision attribute (class) in which a dataset object belongs. This can be defined as a phenotype in Case-Control GWAS. A single instance of the ML dataset can also be an individual in the GWAS population. The training set, test, and validation sets can be denoted as population samples of the genotyped subjects.
Machine Learning
GWAS
(Condition) attribute/feature
Genotype
(Condition) attribute/feature
Covariate
Decision attribute
Phenotype
Instance
Individual from the population described by its condition and decision attributes
Training Set (including test/validation)
Population sample
2 Systematic approach to the study
2.1 Information sources and search strategy
We carried out a search for relevant studies from 2015 to 2021 using online databases such as ScienceDirect and PubMed. This was done to extract relevant research articles that apply machine learning algorithms to genome-wide association studies. The keywords used for the search was done with the query strings “(Machine learning) AND (genome-wide association studies)” both on the PubMed and Science Direct databases. Pubmed retrieved n = 453 articles while Science Direct retrieved n = 378, giving a total number of 827 articles. Duplicate records numbering 20 articles were removed manually while the rest n = 807 were further screened to 122 records after excluding 685 records that did not meet the required criteria as depicted in the model shown in Fig. 1. Finally 22 records were included and reported in the study.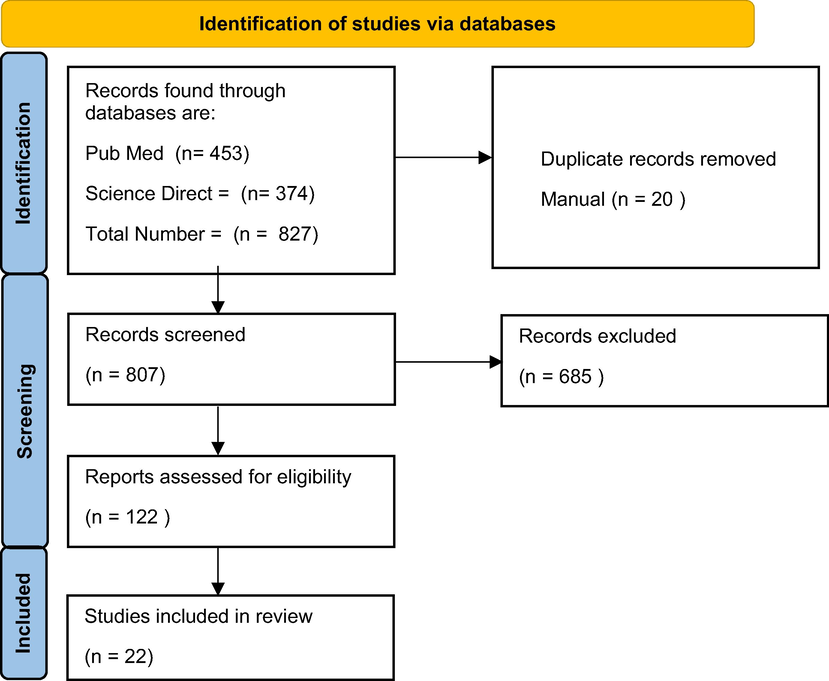
Identification, screening and inclusion of studies.
2.2 Eligibility criteria
The inclusion criteria entails selection for articles that are research articles and not review articles. This is because the focus of the review is for machine learning research articles in genome-wide association studies. Therefore, articles that applied machine learning algorithms in the methodology of the work were included. Additionally, articles that are based on human research were included. The exclusion criteria include the removal of articles that did not relate to the specific research subject in question (Falola et al., 2017). Furthermore, review articles and articles that do not specifically apply ML to their GWASbased work were excluded. The methodology in each of these articles was analysed to identify the specific applications of machine learning to the genome-wide association analysis. From this set of articles, we rigorously reviewed the contributions to the subject of our study with 22 included articles.
3 Algorithms’ classification in GWAS and Post-GWAS
3.1 Supervised machine learning approaches
The supervised machine learning target in this scenario is developing a genotype-phenotype model. This is done by building the patterns from a labelled set of training datasets. The aim is that the model will accurately predict the phenotype in new cases with related genetic backgrounds (Kruppa et al., 2012; Okser et al., 2014). The approaches which have been reviewed include regression, classification, ensemble learning, and neural networks.
3.1.1 Regression
Logistic regression is a popular choice for regression problems. It is sometimes joined with lasso regularization, and this is done after SNP preselection under mild constraints (Romagnoni et al., 2019). This was compared in a study by altering the penalty in the regularization portion of the cost function & preselection constraints. The penalized logistic regression method was applied in classifying Crohn Disease patients with the use of genome-wide genotyping data. Furthermore, the two most commonly applied penalized regression algorithms are LASSO (L1 penalty) and ridge regression (L2 penalty). The algorithms model the phenotype as a linear weighted sum of the genetic variants. This is achieved by the application of a regularization penalty to limit the magnitude of the regression coefficients (Behravan et al., 2018). An et al. (2020) developed a novel algorithm by binning the neighbouring markers (SNPs) according to their Linkage disequilibrium. They then used the LASSO regression method to associate the SNPs with the phenotype. Their method that yielded less type one errors, was faster and more powerful than the other two methods that they were compared against. The two methods are the regular Linear Mixed Model and the SNPs analysed with LASSO alone. Maciukiewicz et al. (2018) performed the standard statistical learning genome-wide logistic regression to find significant variants associated with duloxetine response. They further extracted the best predictors with the help of LASSO regression. In another scenario viz prediction of late genitourinary toxicity after radiotherapy, a preconditioned random forest regression was used. The preconditioning step consists creating a continuous surrogate outcome from original binary outcomes (by logistic regression). This is then accompanied by random forest regression that uses the surrogate outcome as a target for prediction. Five-fold cross validation was also done to test the stability of the model against other baseline models (Lee et al., 2018). A limitation of these regression approaches is that they cannot capture complex epistatic interactions among loci. These are higher level non-linear SNP interactions with disease susceptibility. The method by Zhang et al. (2012) translated the prior knowledge of proteomics and biological pathways to groups of SNPs. Consequently, they applied linear regression that is regularized by group sparse constraint to identify the most predictive SNP groups. Finally, they applied group-LASSO as solution for the regularized linear regression. This approach by Zhang et al. (2012) was applied because of the limitations of the regular Linear Mixed Models that are applied to GWAS data namely- the stringency of the low p-values after Bonferroni correction and abandonment of LD that occurs between neighbouring markers.
3.2 Classification
Mieth et al. (2016) developed an SVM algorithm to predict an unknown phenotype of an unseen genotype x. Consequently, the SNPs that correspond to the greatest values of the SVM weighting are selected and the rest are discarded. A chi-squared test was then performed on the selected SNPs where SNPs with a p-value lesser than the significance threshold were selected. The COMBI method which they developed outperformed Raw P-value thresholding (RPVT). The comparison figure stood at a gain in true positive rate up to 80%. Additionally, their method identified 12 more SNPs. Among these SNPs, 10 have already been replicated in later GWAS and/or meta-analyses. The principle of SVMs is to separate labelled data points into two groups, with a large difference between them (Mittag et al., 2012; Roshan et al., 2011). Mittag et al. (2012) also used the SVM in genetic risk prediction. They developed the method to conduct genome-wide risk profiling for Parkinson’s Disease and Type 1 Diabetes. The algorithm was applied to train models based on GWAS SNP data, which could perform binary classification on a test dataset. Compared to Parkinson’s disease, their method reached satisfactory performance on application to type 1 diabetes datasets. They approached AUC scores in the range of 0.81 to 0.88. For validation they performed within study cross validation and between study validation. Hajiloo et al. (2013) used a simple K-Nearest Neighbours (KNN) learning algorithm to classify breast cancer patients according to their SNPs as having breast cancer or not. They used two strategies for evaluating the classification algorithm viz, Leave-One-Out Cross Validation (LOOCV) strategy and external hold-out (validation). Their LOOCV strategy estimated the accuracy, precision, sensitivity and specificity of the model to be 59.55%, 50.40%, 61.92% & 57.32% respectively.
3.3 Ensemble learning algorithms
The Random Forest is an example of the ensemble machine learning (ML) algorithm. It comprises an ensemble (collection) of decision trees. Each is developed with a bootstrapped subsample of the entire training dataset. Hence, it is anticipated that the ensemble algorithms are well fit for modelling the non-linear biological dependencies apparent in genetic data such as the GWAS SNP data (Lee et al., 2020). This is because the multi-variate and non-linear characteristics of tree-based ensemble learning algorithms have been shown to be a robust analytic tool in detection of genes’ interaction in GWAS (Dorani et al., 2018). Furthermore, ML algorithms have been utilized successfully in GWAS to identify genetic variants which have relatively large consequences in complex disease states. Random forest is one of them (Nguyen et al., 2015). Dorani et al. (2018) utilized random forests and gradient boosting machine to find 44 possibly susceptible SNPs that were ranked most significantly. For the random forests, AUC of 0.84 was achieved when number of predicted variables of 100 and number of trees of 2000 were applied. This means that the algorithm performed best when the number of trees were set to maximum and the number of predictor variables were set to the minimum. Nguyen et al. (2015) focused on a method for choosing informative SNPs with the use of the Random Forest method. They used a new approach in learning Random Forest’s model (ts-RF) with a two-stage quality-based method for subspace selection of SNPs. This method is precisely fit for the high dimensional nature of GWAS data. The fivefold cross-validation was also applied in evaluating the predictive performance of the model on GWAS datasets. Gradient boosting of decision trees was also applied to GWAS datasets. Behravan et al. (2018) developed a SNP selection process with an XGBoost model. Next, there was a repetitive SNP search to get the optimal SNP groups interacting and having a high breast cancer risk potential. This model was developed as a surrogate to the polygenic risk scoring model and included the SVM classifier in its backend for classification of SNPs.
Oh et al. (2017) developed a model called preconditioned random forest regression (PRFR). They converted a binary variable viz toxicity and non-toxicity into a continuous variable. This was achieved with principal component analysis and logistic regression. López et al. (2018) used the Random Forest algorithm to find the most important SNPs related to diabetes. They assigned the weights as values that range between 0 and 1, to each attribute. Lee et al. (2020) applied the preconditioned random forest regression method to predict the risk of contralateral breast cancer. Using a 5-fold cross validation, the PRFR model on 712 SNPs achieved an average AUC of 0.57 (95% confidence interval: 0.57–0.58) on the validation data.
3.4 Neural networks
Liu et al. (2019) proposed an independent deep Convoluted Neural Network (CNN) model to predict phenotypes information from data on SNPs. They were also the first to apply a saliency map deep learning visualization method to select significant biomarkers (SNPs) from their trained model. They were compared against other statistical methods viz Best Linear Unbiased Prediction (BLUP), Bayesian ridge regression (BRR), Bayesian A, and Bayesian Lasso. The design was to associate quantitative traits of soybean with the SNP dataset. Neural networks were applied in a comparative work with other machine learning algorithms. Specifically, Dense neural networks with one fully connected layer, with different numbers of fully connected hidden layer and with varying odd numbers of fully connected hidden layer. They achieved mean AUC scores in the range of 0.80 with the model (Romagnoni et al., 2019). In the neural networks, work done by Romagnoni et al. (2019) noted that an increase in the number of hidden neurons did not significantly increase the performance of the model in terms of classification of cases and controls. The deep mixed model (composed of Convoluted neural network and Long Term Short Term Memory model) was compared against such methods as standard univariate testing & standard linear mixed model (with Benjamini Hochberg procedure) (Wang et al., 2019). However, the results of this method when verified in literature search. seem not verifiable in the GWAS catalogue. The SNP reported by this method are linked to the disease in question-Alzheimer's disease. In predicting the risk for Age-related Macular Degeneration, Neural Networks were also used although it did not show a great advantage over the other methods applied (AUC = 0.82 ∼ 0.83) (Yan et al., 2019).
4 Current trends and application area
4.1 Identification of significant SNPs
A problem with the current methods of GWAS analysis is the dependence on testing each SNP individually. Consequently, this leads to the disregard for any biological correlation structures between the SNPs understudied. These structures are introduced by both population genetics (linkage disequilibrium) and other biological effects (Mieth et al., 2016). The approach by Nguyen et al. (2015) with Random Forest has shown to have effectivity in selecting SNPs that are possibly associated with diseases. Where they succeed, the authors posit that traditional statistical approaches might fail. The machine learning approaches thrive in the high dimensionality problem where the number of sample subjects is notably less than that of the SNPs. Mieth et al. (2016) was also able to combine the Support vector machine classifier with the standard statistical testing, RPVT (Raw P-value thresholding), to identify significant SNPs associated with the WTCCC data. They called their method COMBI. Behravan et al. (2018) developed a gradient tree boosting method to capture the complex non-linear SNP-SNP interactions. Eventually, the method obtains a group of significant SNPs which have high Breast Cancer predictive potential. They also developed a support vector machine classifier formed by the identified SNPs to classify Breast Cancer cases and controls. They achieved approximate precision (mAP) scores of 73, 67, and 69 in classifying Breast Cancer cases and controls on three datasets (individual and merged datasets). Dorani et al. (2018) had used the plink software for prior quality control on the total GWAS data set. They then focused the next stage of their work on the application of two ensemble learning algorithms, random forests, and gradient boosting machine, in identifying the interacting and significant SNPs correlated with colorectal cancer.
4.2 Disease risk assessment and prediction
SNPs from GWAS have been shown to be valuable in estimating the risk of developing the disease phenotype (Kooperberg et al. 2010; Wang et al., 2016). The computational methods are useful to analyse the impact of the identified variants on the disease risk (Vihinen, 2013). In a study on prediction of the risk of an individual sensitivity to radiotherapy, Oh et al. (2017) developed an innovative multi-SNP predictive model based on ML algorithms namely preconditioned random forest regression (PRFR). They concluded that GWAS SNPs can yield useful risk stratification models and identify important biological processes in radiation damage and tissue repair processes. Mittag et al. (2012) applied a SVM model to Parkinson's disease and type 1 diabetes. This was to reveal that apart from effect sizes of risk variants, disease heritability is also important in disease risk prediction. This points to the effects that each disease variant has a role to play in disease risk assessment and prediction. Gaudillo et al. (2019) also built a novel machine learning model to quantify an individual's risk of developing asthma based on SNP data. Maciukiewicz et al. (2018) studied the potential of ML models using GWAS data to predict duloxetine outcomes in major depressive disorders. SVM and classification & regression trees' based models were the models developed and they had a promising sensitivity scores. Oh et al. (2017) have been able to predict the risk of individual radio sensitivity with novel multi-SNP predictive framework dependent on ML algorithms. Their approach outperformed other methods in predicting the risk of erectile dysfunction and late rectal bleeding after prostate cancer radiotherapy. Similarly in the area of radio genomics, Lee et al. (2018) also applied a preconditioned random forest regression method to genome-wide data. This was done to combine the effects of multiple SNPs to predict patients with high late genitourinary toxicity risk. Lee et al. (2020) also predicted the risk of developing radiation associated contralateral breast cancer (RCBC). Wei et al. (2019) used a six SNPs as classifier (LASSO-Cox Regression) and predictor where the latter may assist the current staging system for predicting localised renal cell carcinoma recurrence after surgery. This helps clinicians make better informed decisions about treatment in adjuvant therapy. Yan et al. (2019) also used four Neural networks, lasso regression, support vector machine, and random forest on GWAS data to predict the risk of developing Age-related macular degeneration. It is a neurodegenerative disease with no known cure but can be adequately managed with diagnosis and risk prediction. Fukaya et al. (2018) applied a gradient boosting machine (GBM) model for the agnostic discovery of novel risk factors in varicose vein development.
4.3 Epistasis (non-linear SNPs) detection
The concept of epistasis is simply the interactions among genetic loci. It has been constantly restated as a major factor contributing to the missing heritability in complex diseases (Okser et al., 2013). Seldomly considered is the non-linear interactions between many genetic factors that play an important role in identifying the genetic variations (Dorani and Hu, 2018). Residual feed intake in dairy were studied by Yao et al. (2013) and epistasis was detected and analysis of tree structures was produced with Random Forest. Finally, the 25 most occurring pairwise SNP interactions were reported as potential epistatic interactions. Wu et al. (2010) developed a method called screen and clean to identify liability loci, including SNPs interactions, with the use of LASSO which is a model selection tool for regression in high dimensional data. Recently, a neural network technique was suggested that could theoretically model arbitrary interactions in GWAS between SNPs as an addendum to the mixed models to correct confounder's effect (Wang et al., 2019). The goal was the investigation of marginal epistasis, for which a deep learning-based method was developed to model arbitrary high-level interactions among genetic variants. The deep mixed model was applied to Alzheimer's disease in order to understand the genetic architecture of the disease. The method reported the top 20 SNPs associated with the disease. A deep learning framework for analysis of epistasis and heterogeneity (Li et al., 2018) called DPEH was developed. The 3-stage framework involves detection of epistasis in the first stage, clustering in the second stage, and prediction in the final stage. Prediction deals with developing a diagnosis model for complex diseases.
In the step of epistasis detection, DPEH searches. Candidate epistatic combinations based on multi-objective optimization and chi-square tests are used to filter false-negative epistatic interaction through filtering based on significance levels. Furthermore, Fergus et al. (2020) combined the GWAS quality control and logistic regression with deep learning stacked autoencoders to develop an approach to extract epistatic interactions among SNPs. The complete framework models the effects of epistasis on minor and major SNP interactions. To essentially capture the epistatic interactions a softmax classifier model pre-initialized with the stacked autoencoder was applied. This approach was eventually applied for the classification of preterm birth risk in mothers within the population of African ancestry. Their model achieved a Sensitivity of 0.96, Specificity of 0.88, and AUC score of 0.97.
A summary of existing machine learning methods and their inferences as applicable to Genome-wide association studies are listed in Table 2.
Author(s)
Method
Result/Inference
Maciukiewicz et al. (2018)
SVM, LASSO, and SNPs data to predict major depressive disorder and adverse drug response (duloxetine)
The models developed had a promising sensitivity however the specificity remain modest in the best case. The best accuracy was 0.66 and sensitivity was 0.70
Behravan et al. (2018)
XGOOST was used to select SNPs in a breast cancer risk prediction task. SVM was then used to distinguish breast cancer cases between cases and healthy controls.
This approach yielded mean average precision of 72.66, 67.24 and 69.25 in classifying breast cancer cases and controls. This was in KBCP, OBCS and merged datasets respectively.
Dorani et al. (2018)
Random forest and gradient boosting machine were applied to search for risk susceptibility SNPs associated with colorectal cancer (CRC).
TuRF feature selection method was applied on all 186,251 SNPs and filtered the top 2798 SNPs. They identified 44 of the most important SNPs with the ML algorithms.
Mieth et al. (2016)
An SVM and SNP selection step (COMBI) was applied on the datasets in order to find the candidate SNPs that are most predictive of the phenotype.
78 SNPs were found to be significant with standard univariate GWAS analysis. 46 with the COMBI method The method also outperformed the RPVT approach for different type 1 error levels.
Romagnoni et al. (2019)
Penalized logistic regression (LR), gradient boosted trees (GBT) and artificial neural networks (ANN) were applied comparatively on a case control dataset.
The maximum AUC values obtained by LR, GBT or NN are in the range of 0.80.
5 Research issues and open problems
5.1 Challenges in GWAS
Traditional statistical methods that apply linear mixed models and logistic regression in case control datasets are not well suited for detecting interactive and non-linear effects. This is especially when there is a large number of predictors. Additionally, when higher level and non-linear interactions are present. The linear modelling method used in GWAS frequently considers only one SNP, thereby ignoring the environmental and genetic context (Moore et al., 2010). It is well known that one of the most significant difficulties to be solved in the detection of GWAS-associated SNPs is in the modelling of complex interactions like higher-level non-linear interactions between SNPs and a given biological phenotype (Behravan et al., 2018). Population stratification causes false positive outcomes in genetic association studies, especially in case-control studies (Menting et al., 2014). Until this moment, genomic data has been explored on the premise of single-locus statistical analyses. The approach is able to identify variant with large effects within a population. However, it is unable to capture variants with smaller effects which may have larger effect sizes when in joint interaction with other SNPS. Detection of SNP interactions remains a significant problem because of the high-dimensionality of genomic data, including the GWAS datasets. This is due to such characteristics as biomolecular complexity, lack of marginal effects, missing heritability, and the limits of computational capacities (Gusareva et al., 2014; Padyukov, 2013; Uppu and Krishna, 2018).
5.2 African genetic diversity
There is relative low number of genetic studies that involve people of African ancestry (Benafif et al., 2018; Gurdasani et al., 2015; Mulder et al., 2018; Radouani et al., 2020). For instance neurogenetic studies of people of African ancestry only account for 11.1 percent in the GWAS catalogue (Quansah and McGregor, 2018). Popejoy and Fullerton (2016) further deliberated on the gross underrepresentation of people of non-European ancestry in Genome-wide Association studies. H3Africa projects have continuously played a role in advancing research bioinformatic capacity and output from African nations (Mulder et al., 2017). African and admixed populations have a more complex haplotype block structure and, as such, will benefit from a broader reference dataset containing more genetic diversity (Schurz et al., 2019). African populations are also characterized by extensive population structure and lower LD among loci relative to non-African population (Campbell and Tishkoff, 2008). Consequently, a larger number of SNPs is required in order to capture the genetic variation within the African genome. It is known that the African population is the most genetically diverse in the world (Bentley et al., 2020). Gurdasani et al. (2015) further reiterated that this haplotype diversity would affect the design of genomics studies. The diversity of the African genome (Choudhury et al., 2018; Ramsay et al., 2011) gives more reason to develop methodologies that can capture non-linear interaction of SNPs, such as the machine learning algorithms described in Section 3.
5.3 Challenges with the adoption of Machine learning in GWAS
Romagnoni et al. (2019) also stated that after training the model, the performance of the model should be estimated on a completely different dataset. This is done in a bid toward evaluation of the ability proposed algorithm to be generalizable. This is similar to the case of having a validation dataset. This is tedious because of the large amount of data that would be generated and the complexity of the entire work. The major weakness of the machine learning model in GWAS is the difficulty in application of the algorithm and difficulty in interpretation of the underlying genetic effects from the results (Ho et al., 2019). The nature of the GWAS dataset is also challenging to pre-process before the eventual model is built. Grinberg et al. (2020) suggested that there is room for the development of new machine learning approaches that make better use of prior knowledge of population structure. They further proposed better collaboration between the machine learning and statistical genetics communities. This is because machine learning suffers from a source of technically interesting and societally important problems which could be gained from the latter.
6 Future trends
Many quantitative traits have a polygenic nature and this makes such a multiple marker association study a better choice than the single marker scanning approach. Even though the single marker scanning approach is still used currently (An et al., 2020). Furthermore, there will be development of more machine learning approaches to handle the high dimensional data generated in GWAS. It is unavoidable because these approaches are better able to capture the missing heritability of these data- including non-linear SNP interactions and epistasis detection. These methods will be able to better prioritize SNPs and perform risk prediction from genome-wide association data. These approaches could also be applied in quantitative traits of plants and animals. Thus, leading to genetically superior species with improved crop yield, milk quality, nutrient quality etc. In the area of precision medicine. In this case, a patient's SNP data could be applied in prediction of an individual's disease risk which could inform decisions such as dosing and treatment regimen. These SNPs could be better fit for post-GWAS analysis and eventually integration of other omics data sources. These will ultimately lead to better insight into disease biology.
7 Conclusion
Machine Learning is a technique widely applied in many application areas, and Biology, Healthcare & Medicine happens to be one of them. A PubMed search saw a progressive increase in the total number of research articles with the keywords “machine learning” and “genome-wide association studies” The high dimensionality characteristic of GWAS data makes it amenable to machine learning algorithms. These algorithms application in GWAS tends to support the understanding of the effects of population genetic variant while highlighting attendant challenges and open problems. It is true that this work has also shown that classification, regression, ensemble, and neural networks have been applied to GWAS as comprehensively discussed giving understanding to African genetic diversity. Machine learning has been shown to have application areas to many areas of genome-wide association studies, including, computational pipeline development and integration with other omics datasets. The studies have shown that ML approaches and regular statistical approaches can help in identification of significant SNPs, detection of epistasis and disease risk prediction. This will be highly beneficial in the design and execution of genetic studies. Finally, it is expected that machine learning algorithms will become a mainstay in the computational pipeline of genome-wide association studies.
Acknowledgements
We would like to acknowledge Covenant University and Covenant University Centre for Research and Innovation’s support for the funding of the publication of this work.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Genome-wide association studies using binned genotypes. Heredity (Edinb).. 2020;124:288-298.
- [CrossRef] [Google Scholar]
- Machine learning identifies interacting genetic variants contributing to breast cancer risk: A case study in Finnish cases and controls. Sci. Rep.. 2018;8:13149.
- [CrossRef] [Google Scholar]
- A review of prostate cancer Genome-Wide Association Studies (GWAS) Cancer Epidemiol. Biomarkers Prev. 2018
- [CrossRef] [Google Scholar]
- Bentley, A.R., Callier, S.L., Rotimi, C.N., 2020. Evaluating the promise of inclusion of African ancestry populations in genomics. npj Genomic Med. https://doi.org/10.1038/s41525-019-0111-x
- African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu. Rev. Genomics Hum. Genet.. 2008;9:403-433.
- [CrossRef] [Google Scholar]
- African genetic diversity provides novel insights into evolutionary history and local adaptations. Hum. Mol. Genet. 2018
- [CrossRef] [Google Scholar]
- Feature Selection for Detecting Gene-Gene Interactions in Genome-Wide Association Studies. In: Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics). Springer Verlag; 2018. p. :33-46.
- [CrossRef] [Google Scholar]
- Ensemble learning for detecting gene-gene interactions in colorectal cancer. PeerJ. 2018;6:e5854
- [CrossRef] [Google Scholar]
- Analyzing a single nucleotide polymorphism in schizophrenia: a meta-analysis approach. Neuropsychiatr. Dis. Treat.. 2017;13:2243-2250.
- [CrossRef] [Google Scholar]
- Utilizing deep learning and genome wide association studies for epistatic-driven preterm birth classification in African-American Women. IEEE/ACM Trans. Comput. Biol. Bioinforma.. 2020;17:668-678.
- [CrossRef] [Google Scholar]
- Clinical and genetic determinants of varicose veins: Prospective, community-based study of ≈500 000 individuals. Circulation. 2018;138:2869-2880.
- [CrossRef] [Google Scholar]
- Machine learning approach to single nucleotide polymorphism-based asthma prediction. PLoS One. 2019;14:e0225574
- [CrossRef] [Google Scholar]
- An evaluation of machine-learning for predicting phenotype: studies in yeast, rice, and wheat. Mach. Learn.. 2020;109:251-277.
- [CrossRef] [Google Scholar]
- The African genome variation project shapes medical genetics in Africa. Nature. 2015;517:327-332.
- [CrossRef] [Google Scholar]
- Genome-wide association interaction analysis for Alzheimer’s disease. Neurobiol. Aging. 2014;35:2436-2443.
- [CrossRef] [Google Scholar]
- Breast cancer prediction using genome wide single nucleotide polymorphism data. BMC Bioinformatics. 2013;14:S3.
- [CrossRef] [Google Scholar]
- Machine learning SNP based prediction for precision medicine. Front. Genet. 2019
- [CrossRef] [Google Scholar]
- Risk prediction using genome-wide association studies. Genet. Epidemiol.. 2010;34:643-652.
- [CrossRef] [Google Scholar]
- Risk estimation and risk prediction using machine-learning methods. Hum. Genet. 2012
- [CrossRef] [Google Scholar]
- Machine learning on a genome-wide association study to predict late genitourinary toxicity after prostate radiation therapy. Int. J. Radiat. Oncol. Biol. Phys.. 2018;101:128-135.
- [CrossRef] [Google Scholar]
- Machine learning on genome-wide association studies to predict the risk of radiation-associated contralateral breast cancer in the WECARE Study. PLoS One. 2020;15:e0226157
- [CrossRef] [Google Scholar]
- Heterogeneity analysis and diagnosis of complex diseases based on deep learning method. Sci. Rep.. 2018;8:6155.
- [CrossRef] [Google Scholar]
- Phenotype prediction and genome-wide association study using deep convolutional neural network of soybean. Front. Genet.. 2019;10:1091.
- [CrossRef] [Google Scholar]
- Single nucleotide polymorphism relevance learning with random forests for type 2 diabetes risk prediction. Artif. Intell. Med.. 2018;85:43-49.
- [CrossRef] [Google Scholar]
- GWAS-based machine learning approach to predict duloxetine response in major depressive disorder. J. Psychiatr. Res.. 2018;99:62-68.
- [CrossRef] [Google Scholar]
- Protective hinge in insulin opens to enable its receptor engagement. Proc. Natl. Acad. Sci. U S A. 2014;111:E3395-E3404.
- [CrossRef] [Google Scholar]
- Combining multiple hypothesis testing with machine learning increases the statistical power of genome-wide association studies. Sci. Rep.. 2016;6:36671.
- [CrossRef] [Google Scholar]
- Use of support vector machines for disease risk prediction in genome-wide association studies: Concerns and opportunities. Hum. Mutat.. 2012;33:1708-1718.
- [CrossRef] [Google Scholar]
- Bioinformatics challenges for genome-wide association studies. Bioinformatics 2010
- [CrossRef] [Google Scholar]
- Mulder, N., Abimiku, A., Adebamowo, S.N., de Vries, J., Matimba, A., Olowoyo, P., Ramsay, M., Skelton, M., Stein, D.J., 2018. H3Africa: Current perspectives. Pharmgenomics. Pers. Med. https://doi.org/10.2147/PGPM.S141546.
- Development of Bioinformatics Infrastructure for Genomics Research. Glob. Heart. 2017;12:91-98.
- [CrossRef] [Google Scholar]
- Genome-wide association data classification and SNPs selection using two-stage quality-based random forests. BMC Genomics. 2015;16:S5.
- [CrossRef] [Google Scholar]
- Computational methods using genome-wide association studies to predict radiotherapy complications and to identify correlative molecular processes. Sci. Rep.. 2017;7:1-10.
- [CrossRef] [Google Scholar]
- Regularized machine learning in the genetic prediction of complex traits. PLoS Genet.. 2014;10
- [CrossRef] [Google Scholar]
- Genetic variants and their interactions in disease risk prediction - Machine learning and network perspectives. BioData Min 2013
- [CrossRef] [Google Scholar]
- Between the Lines of Genetic Code: Genetic Interactions in Understanding Disease and Complex Phenotypes. Elsevier Science; 2013.
- Towards diversity in genomics: The emergence of neurogenomics in Africa? Genomics 2018
- [CrossRef] [Google Scholar]
- A review of clinical pharmacogenetics Studies in African populations. Per. Med.. 2020;17:155-170.
- [CrossRef] [Google Scholar]
- Africa: The next frontier for human disease gene discovery? Hum. Mol. Genet.. 2011;20:214-220.
- [CrossRef] [Google Scholar]
- A. Romagnoni S. Jégou K. Van Steen G. Wainrib J.P. Hugot L. Peyrin-Biroulet M. Chamaillard J.F. Colombel M. Cottone M. D’Amato R. D’Incà J. Halfvarson P. Henderson A. Karban N.A. Kennedy M.A. Khan M. Lémann A. Levine D. Massey M. Milla S.M.E. Ng I. Oikonomou H. Peeters D.D. Proctor J.F. Rahier P. Rutgeerts F. Seibold L. Stronati K.M. Taylor L. Törkvist K. Ublick J. Van Limbergen V. Gossum V. Andre M.H., Zhang, H., Zhang, W., Andrews, J.M., Bampton, P.A., Barclay, M., Florin, T.H., Gearry, R., Krishnaprasad, K., Lawrance, I.C., Mahy, G., Montgomery, G.W., Radford-Smith, G., Roberts, R.L., Simms, L.A., Hanigan, K., Croft, A., Amininijad, L., Cleynen, I., Dewit, O., Franchimont, D., Georges, M., Laukens, D., Peeters, H., Rahier, J.F., Rutgeerts, P., Theatre, E., Van Gossum, André, Vermeire, S., Aumais, G., Baidoo, L., Barrie, A.M., Beck, K., Bernard, E.J., Binion, D.G., Bitton, A., Brant, S.R., Cho, J.H., Cohen, A., Croitoru, K., Daly, M.J., Datta, L.W., Deslandres, C., Duerr, R.H., Dutridge, D., Ferguson, J., Fultz, J., Goyette, P., Greenberg, G.R., Haritunians, T., Jobin, G., Katz, S., Lahaie, R.G., McGovern, D.P., Nelson, L., Ng, S.M., Ning, K., Oikonomou, I., Paré, P., Proctor, D.D., Regueiro, M.D., Rioux, J.D., Ruggiero, E., Schumm, L.P., Schwartz, M., Scott, Regan, Sharma, Y., Silverberg, M.S., Spears, D., Steinhart, A.H., Stempak, J.M., Swoger, J.M., Tsagarelis, C., Zhang, W., Zhang, C., Zhao, H., Aerts, J., Ahmad, T., Arbury, H., Attwood, A., Auton, A., Ball, S.G., Balmforth, A.J., Barnes, C., Barrett, J.C., Barroso, I., Barton, A., Bennett, A.J., Bhaskar, S., Blaszczyk, K., Bowes, J., Brand, O.J., Braund, P.S., Bredin, F., Breen, G., Brown, M.J., Bruce, I.N., Bull, J., Burren, O.S., Burton, J., Byrnes, J., Caesar, S., Cardin, N., Clee, C.M., Coffey, A.J., MC Connell, J., Conrad, D.F., Cooper, J.D., Dominiczak, A.F., Downes, K., Drummond, H.E., Dudakia, D., Dunham, A., Ebbs, B., Eccles, D., Edkins, S., Edwards, C., Elliot, A., Emery, P., Evans, D.M., Evans, G., Eyre, S., Farmer, A., Ferrier, I.N., Flynn, E., Forbes, A., Forty, L., Franklyn, J.A., Frayling, T.M., Freathy, R.M., Giannoulatou, E., Gibbs, P., Gilbert, P., Gordon-Smith, K., Gray, E., Green, E., Groves, C.J., Grozeva, D., Gwilliam, R., Hall, A., Hammond, N., Hardy, M., Harrison, P., Hassanali, N., Hebaishi, H., Hines, S., Hinks, A., Hitman, G.A., Hocking, L., Holmes, C., Howard, E., Howard, P., Howson, J.M.M., Hughes, D., Hunt, S., Isaacs, J.D., Jain, M., Jewell, D.P., Johnson, T., Jolley, J.D., Jones, I.R., Jones, L.A., Kirov, G., Langford, C.F., Lango-Allen, H., Lathrop, G.M., Lee, J., Lee, K.L., Lees, C., Lewis, K., Lindgren, C.M., Maisuria-Armer, M., Maller, J., Mansfield, J., Marchini, J.L., Martin, P., Massey, D.C., McArdle, W.L., McGuffin, P., McLay, K.E., McVean, G., Mentzer, A., Mimmack, M.L., Morgan, A.E., Morris, A.P., Mowat, C., Munroe, P.B., Myers, S., Newman, W., Nimmo, E.R., O’Donovan, M.C., Onipinla, A., Ovington, N.R., Owen, M.J., Palin, K., Palotie, A., Parnell, K., Pearson, R., Pernet, D., Perry, J.R., Phillips, A., Plagnol, V., Prescott, N.J., Prokopenko, I., Quail, M.A., Rafelt, S., Rayner, N.W., Reid, D.M., Renwick, A., Ring, S.M., Robertson, N., Robson, S., Russell, E., Clair, D.S., Sambrook, J.G., Sanderson, J.D., Sawcer, S.J., Schuilenburg, H., Scott, C.E., Scott, Richard, Seal, S., Shaw-Hawkins, S., Shields, B.M., Simmonds, M.J., Smyth, D.J., Somaskantharajah, E., Spanova, K., Steer, S., Stephens, J., Stevens, H.E., Stirrups, K., Stone, M.A., Strachan, D.P., Su, Z., Symmons, D.P.M., Thompson, J.R., Thomson, W., Tobin, M.D., Travers, M.E., Turnbull, C., Vukcevic, D., Wain, L. V., Walker, M., Walker, N.M., Wallace, C., Warren-Perry, M., Watkins, N.A., Webster, J., Weedon, M.N., Wilson, A.G., Woodburn, M., Wordsworth, B.P., Yau, C., Young, A.H., Zeggini, E., Brown, M.A., Burton, P.R., Caulfield, M.J., Compston, A., Farrall, M., Gough, S.C.L., Hall, A.S., Hattersley, A.T., Hill, A.V.S., Mathew, C.G., Pembrey, M., Satsangi, J., Stratton, M.R., Worthington, J., Hurles, M.E., Duncanson, A., Ouwehand, W.H., Parkes, M., Rahman, N., Todd, J.A., Samani, N.J., Kwiatkowski, D.P., McCarthy, M.I., Craddock, N., Deloukas, P., Donnelly, P., Blackwell, J.M., Bramon, E., Casas, J.P., Corvin, A., Jankowski, J., Markus, H.S., Palmer, C.N., Plomin, R., Rautanen, A., Trembath, R.C., Viswanathan, A.C., Wood, N.W., Spencer, C.C.A., Band, G., Bellenguez, C., Freeman, C., Hellenthal, G., Giannoulatou, E., Pirinen, M., Pearson, R., Strange, A., Blackburn, H., Bumpstead, S.J., Dronov, S., Gillman, M., Jayakumar, A., McCann, O.T., Liddle, J., Potter, S.C., Ravindrarajah, R., Ricketts, M., Waller, M., Weston, P., Widaa, S., Whittaker, P., Comparative performances of machine learning methods for classifying Crohn Disease patients using genome-wide genotyping data Sci. Rep. 9 2019 1 18 10.1038/s41598-019-46649-z
- Roshan, U., Chikkagoudar, S., Wei, Z., Wang, K., Hakonarson, H., 2011. Ranking causal variants and associated regions in genome-wide association studies by the support vector machine and random forest. Nucleic Acids Res. 39, e62–e62. https://doi.org/10.1093/nar/gkr064.
- Evaluating the accuracy of imputation methods in a five-way admixed population. Front. Genet.. 2019;10:34.
- [CrossRef] [Google Scholar]
- Machine learning in genome-wide association studies. Genet. Epidemiol.. 2009;33:S51-S57.
- [CrossRef] [Google Scholar]
- A deep hybrid model to detect multi-locus interacting SNPs in the presence of noise. Int. J. Med. Inform.. 2018;119:134-151.
- [CrossRef] [Google Scholar]
- Guidelines for reporting and using prediction tools for genetic variation Analysis. Hum. Mutat.. 2013;34:275-282.
- [CrossRef] [Google Scholar]
- Deep mixed model for marginal epistasis detection and population stratification correction in genome-wide association studies. BMC Bioinformatics. 2019;20:656.
- [CrossRef] [Google Scholar]
- Genetic markers of type 2 diabetes: Progress in genome-wide association studies and clinical application for risk prediction. J. Diabetes. 2016;8:24-35.
- [CrossRef] [Google Scholar]
- Random forests on Hadoop for genome-wide association studies of multivariate neuroimaging phenotypes. BMC Bioinformatics. 2013;14:S6.
- [CrossRef] [Google Scholar]
- Predictive value of single-nucleotide polymorphism signature for recurrence in localised renal cell carcinoma: a retrospective analysis and multicentre validation study. Lancet Oncol.. 2019;20:591-600.
- [CrossRef] [Google Scholar]
- Screen and clean: a tool for identifying interactions in genome-wide association studies. Genet. Epidemiol.. 2010;34:n/a-n/a.
- [CrossRef] [Google Scholar]
- Yan, Q., Jiang, Y., Huang, H., Swaroop, A., Chew, E., Weeks, D., Chen, W., Ding, Y., 2019. GWAS-based Machine Learning for Prediction of Age-Related Macular Degeneration Risk. medRxiv 19006155. https://doi.org/10.1101/19006155.
- Random Forests approach for identifying additive and epistatic single nucleotide polymorphisms associated with residual feed intake in dairy cattle. J. Dairy Sci.. 2013;96:6716-6729.
- [CrossRef] [Google Scholar]
- Zhang, Z., Xu, Y., Liu, J., Kwoh, C.K., 2012. Identify predictive SNP groups in genome wide association study: A sparse learning approach, in: Procedia Computer Science. Elsevier B.V., pp. 107–114. https://doi.org/10.1016/j.procs.2012.09.012




