

Translate this page into:
Investigation and comparative study of the quantum molecular descriptors derived from the theoretical modeling and Monte Carlo simulation of two new macromolecular polyepoxide architectures TGEEBA and HGEMDA
⁎Corresponding authors. r.hsissou@gmail.com (R. Hsissou), azarrouk@gmail.com (A. Zarrouk)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Our objective is to compare the performance of the quantum parameters of two new synthesized multifunctional polymeric architectures: triglycidyl ether ethylene of bisphenol A (TGEEBA) and hexaglycidyl ethylene of methylene dianiline (HDEMDA). Calculations have been performed with Gaussian software package and the prediction of the quantum molecular descriptors of the two new macromolecular matrices are namely: the energy of the highest occupied molecular orbital (EHOMO), the energy of the lowest molecular orbital (ELUMO), the ionization potanial (IP), the electron affinity (EA), gap energy (ΔE), softness (σ), electronegativity (χ), hardness (ƞ), the electrophilic character (ω) and the function of the transferred electrons of coating on the surface of the metal (ΔN), etc. These were calculated by the functional density theory method (DFT) with 6–311 G (d, p) basis sets. The local reactivity of two polymers has been studied through the Fukui indices. Indeed, we conclude that the matrix HGEMDA has a low value of the adsorption energy than that of the TGEEBA, indicating that this polymer has a high adsorption capacity on the metal surface. Finally, the results obtained by the Monte Carlo simulation are in very good agreement with the data of the theory of the DFT.
Keywords
Polymer
Architecture
DFT
Local reactivity
Fukui indices
Monte Carlo

1 Introduction
The usage of new the multifunctional polyepoxide polymer architectures synthesized in the laboratory with interesting macromolecular properties is a field of application in the protection of coating corrosion (Hsissou et al., 2015; Hsissou et al., 2017a,b,c; Bekhta et al., 2016). Their counterparts have of course applications in the various domains (Hsissou et al., 2017a,b,c). The ability to predict the properties of the new material is an approach that would accelerate the development process while the field of corrosion related to inhibition, coating and paint which are evaluated by electrochemical studies on the metal support remains one of the most sought after applications for organic polymers (Hsissou et al., 2017a,b,c). The study of the quantum molecular descriptors predicts their effectiveness of adhesion on the substrate. Many methods are proposed to predict the coating behavior of polymer systems, which include empirical equations (Venditti and Gillham, 1997; Delrio et al., 1997), molecular dynamics simulations (Tan and Rode, 1996; Tsige and Taylor, 2002), semi-empirical methods (Gumen et al., 2001) and mathematical tools, including neural networks (Ulmer Ila et al., 1998; Jurs and Mattioni, 2002), theory of fuzzy sets (Joyce et al., 1995) and graph of theoretical indices (Bicerano, 1996; Garcia and Julian, 2002).
We studied in this work the correlation of the molecular properties of two new synthesized polyepoxide prepolymers: triglycidyl ether ethylene of bisphenol A (TGEEBA) and hexaglycidyl ethylene of methylene dianiline (HDEMDA) with the calculations of the quantum molecular descriptors.
2 Material and methods
2.1 Macromolecular matrices
We studied in this work the calculation of the quantum molecular descriptors of two new tri and hexa-functional polyepoxide polymer synthesized in the laboratory: triglycidyl ether ethylene of bisphenol A (TGEEBA) (Hsissou et al., 2016) and hexaglycidyl ethylene of methylene dianiline (HDEMDA) (Hsissou et al., 2017a,b,c). The latter are shown in Figs. 1 and 2.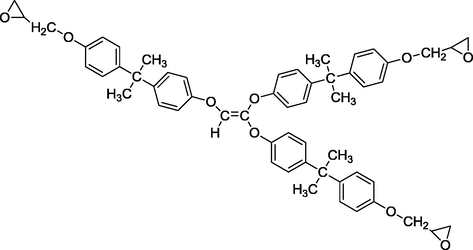
Semi-developed structure of triglycidyl ether ethylene of bisphenol A.
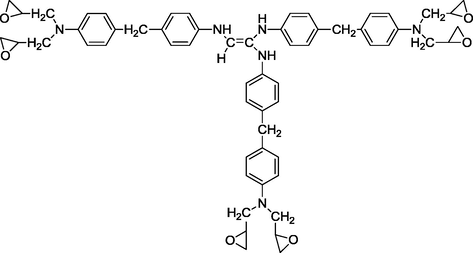
Semi-developed structure of hexaglycidyl ethylene of methylene dianiline.
2.2 Calculation method
Quantum molecular descriptors calculations have been widely used to study reaction mechanisms (Ziraoui et al., 2010) and they have also approved to be a very interesting tool to study the properties of molecules (Tao et al., 2010; Hsissou et al., 2018). It has been shown that the physical properties can be related to the molecular structure.
Therefore, we studied the relationship between the quantum molecular descriptors of two matrices which were calculated by the DFT method on the 6-311G (d, p) basis sets (Ansari and Quraishi, 2015; Neese, 2012; Becke, 1986). All these calculations have been performed with Gaussian (03) software package. The exchange–correlation was treated using hybrid, B3LYP functionals. A full optimization was performed using the 6-311G (d,p) basis sets. This basis set is well known to provide accurate geometries and electronic properties for a wide range of organic compounds. Default optimization criteria (Max Force = 0.00045, RMS Force = 0.0003, Max Displacement = 0.0018 and RMS Displacement = 0.0012) was adopted. Frequency analysis was performed to ensure that the calculated structures are at a minimum point on the potential energy surface (without imaginary frequency).
The calculated quantum molecular descriptors are the energy of the highest occupied molecular orbital (EHOMO), and the energy of the lowest unoccupied molecular orbital (ELUMO) (Neese, 2012). These molecular orbits EHOMO and ELUMO of the molecule are related to ionization energy (IE) and electron affinity (EA) (Lee et al., 1988; Saha et al., 2014).
The gap energy is the difference between the energy of the lowest unoccupied molecular orbital (ELUMO) and the energy of the highest occupied molecular orbital (EHOMO).
The absolute chemical hardness (η) can be evaluated by the following equation (Becke, 1986).
The chemical softness (σ), which describes the ability of an atom or a group of atoms to accept electrons, is calculated according to the following equation (Lee et al., 1988).
The electronegativity (χ) of coating behavior is calculated by the following equation [28].
Furthermore, the electrophilic character ω is a reactivity descriptor allowing a quantitative classification of the electrophilic nature of a compound within a relative scale. We proposed ω as a measure of the lowering of the maximum energy due to electron flows between the donor and the acceptor, in which ω is defined by the following relation (Saha et al., 2014).
The number of transferred electrons (ΔN) was calculated according to the quantum chemical method by the following equation (El Janati et al., 2016)
The local reactivity of the optimized polymers was carried out by the Fukui indices. This study indicates the region of reactivity an atom or the set of atoms, which are responsible for the nucleophilic and electrophilic attack of each of these atoms in the tested molecules.
2.3 Detail of the use of the Monte Carlo simulation
The interaction between the investigated inhibitors and Fe (1 1 0) plane surface was carried out using Monte Carlo simulations. The adsorption locator code implemented in the Material Studio 7.0 was adopted in this simulation. The computation was performed using condensed phase optimized molecular fields for atomic simulation studies (COMPASS). The simulation MC was executed for the system that contains the polymer studied and the iron surface (1 1 0), this surface is considered the most stable in comparison with the surfaces of Fe (1 0 0) and Fe (1 1 1) (Guo et al., 2014). The interaction between the surface of Fe (1 1 0) and the new resin studied was performed in a simulation box size of (20.12 × 20.12 × 35.8 Å). The Fe (1 1 0) plane was then enlarged to a super-cell (6.6) with periodic boundary conditions. A vacuum plate of 30 Å was introduced on the surface Fe (1 1 0). The simulation of the corrosion inhibitor molecules on Fe (1 1 0) surface was carried out in order to locate the low energy adsorption sites of the potential corrosion inhibitors on Fe surface. Smart algorithm was adopted for the optimization based on simulated annealing procedures with convergence criteria quality set to fine. The ewald & group method (ewald accuracy: 1.0 × 10−5 kcal/mol) was applied for the electrostatic interaction, and atom based method (cutoff distance: 1.85 nm) for the van der Waals interaction.
3 Results and discussion
3.1 Quantum computation
The experimental results which concern the behavior of the coating of E24 carbon steel in a marine environment obtained by the stationary and transient electrochemical studies showed simultaneously that the protection efficiency of the metal by the different formulations E1 ((TGEEBA/MDA) and (HGEMDA/MDA)) E2 ((TGEEBA/MDA/PN) and (HGEMDA/MDA/PN)) from the macromolecular structures of the new synthesized polyepoxide polymeric architectures: triglycidyl ether ethylene of bisphenol A (TGEEBA) and hexaglycidyl ethylene of methylene dianiline (HGEMDA) are very good.
In order to confirm the adhesion sites of the epoxy polymers which are responsible for the experimental aquis results, reflecting the good adhesion of our macromolecular tri and hexa-functional epoxy matrices, composed of six aromatic nuclei on the one hand and one ethylenic radical on the other hand, we carried out the comparative study of the calculations of quantum molecular descriptors by Gaussian software 03W.
Likewise, to perform the calculations of the quantum molecular descriptors of epoxy resins, several descriptors have been studied on the electronic steric basis. The calculation of the different descriptors was carried out by the DFT method on the 6-311G (d, p) basis sets. Therefore, the quantum molecular descriptors were calculated and analyzed in order to explain the reactivity of the electronegative and electropositive sites of the TGEEBA and HGEMDA molecules which are grouped in Table 1. The optimized geometric structures and the density distributions of the EHOMO and ELUMO electrons for these polymers are thus presented in Figs. 3–9.
Matrices
TGEEBA
HGEMDA
Load
Neutral
Negative load
Positive load
Neutral
Negative load
Positive load
EHOMO (eV)
−5.876
−7.243
−7.21
−6.499
−6.499
−6.344
ELUMO (eV)
−2.536
−4.168
−5.003
−4.625
−4.631
−4.789
ΔE gap (eV)
3.340
3.075
2.207
1.874
1.868
1.555
I (eV)
5.876
7.243
7.210
6.499
6.499
6.344
A (eV)
2.536
4.168
5.003
4.625
4.631
4.789
μ (Debye)
7.777
37.450
21.259
3.143
8.438
9.122
η (eV)
1.670
1.537
1.103
0.937
0.934
0.777
σ (eV−1)
0.598
0.650
0.906
1.067
1.070
1.287
χ (eV)
4.206
5.705
6.106
5.562
5.565
5.566
ω (eV)
5.296
10.588
16.90
16.507
16.578
19.935
ΔN (eV)
0.836
0.421
0.405
0.767
0.768
0.922
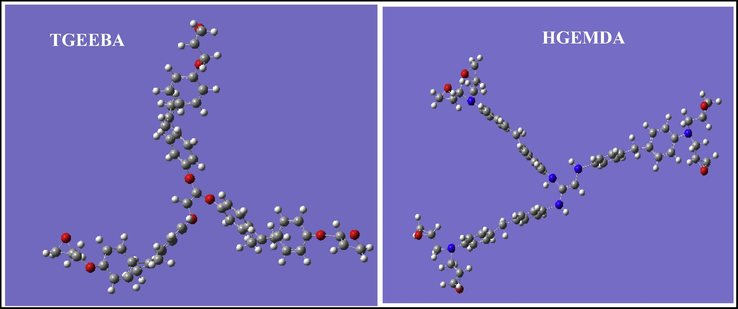
Optimiszation structures of tow polymers TGEEBA and HGEMDA.
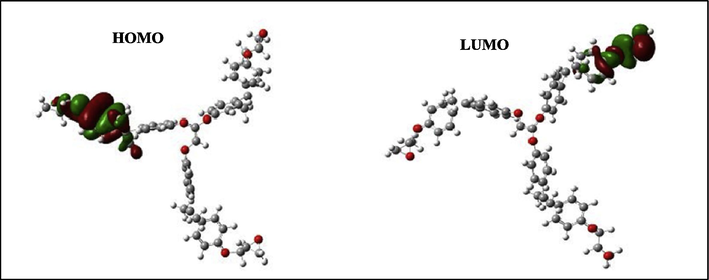
HOMO and LUMO orbitals of the neutral TGEEBA.
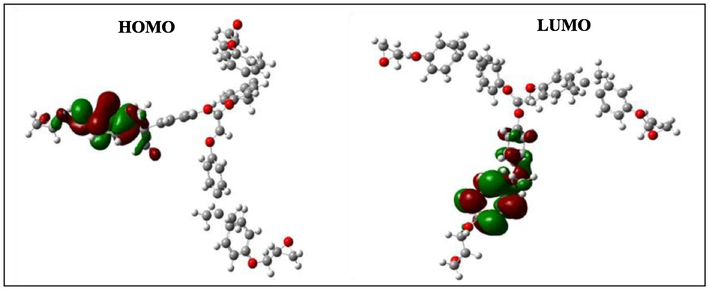
HOMO and LUMO orbitals of the negatively charged TGEEBA.
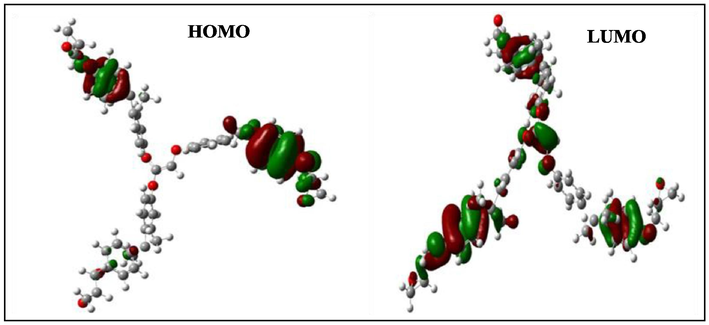
HOMO and LUMO orbits of the positively charged TGEEBA.
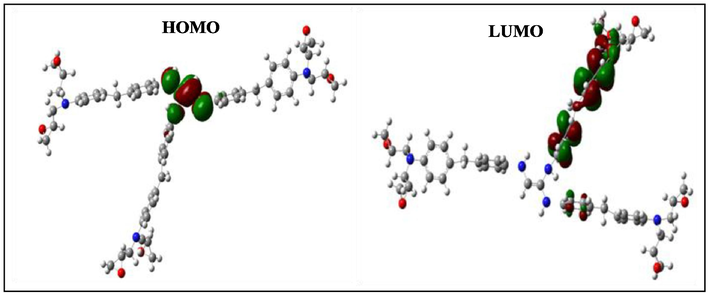
HOMO and LUMO orbitals of the neutral HGEMDA.
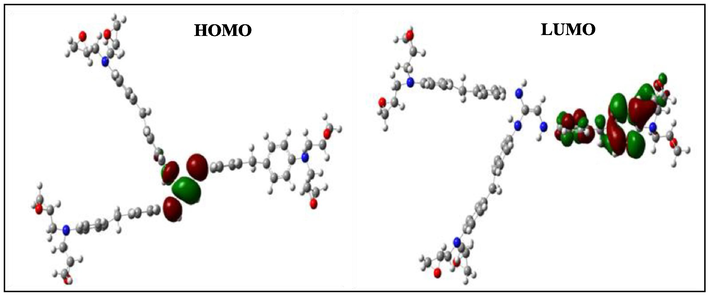
HOMO and LUMO orbitals of the negatively charged HGEMDA.
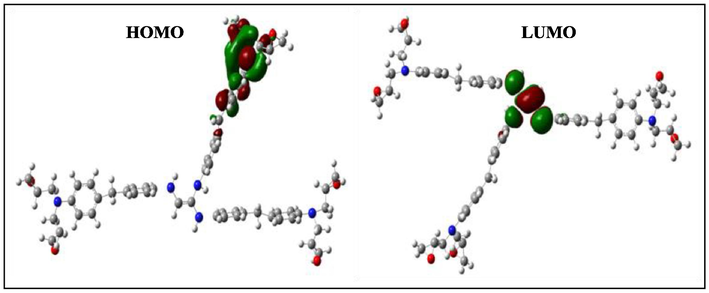
HOMO and LUMO orbitals of the positively charged HGEMDA.
According to Figs. 4–9, we have observed that the electronic density (HOMO) is located on the aromatic ring surface and the epoxy group for the TGEEBA matrix (neutral, negatively charged and positively charged) and on the ethylenic ring for the HGEMDA (neutral and negatively charged) matrix thus on the aromatic ring surface and the epoxy group for the positively charged HGEMDA matrix. On the other hand, the electronic densities (LUMO) are located on the epoxy ring surface for the neutral TGEEBA matrix, on the surface of the aromatic ring for the negatively charged TGEEBA, on the aromatic ring surface and the ethylenic nucleus for the positively charged TGEEBA; and on the surface of the aromatic and epoxy rings for the HGEMDA (neutral and negatively charged) matrix, and finally on the ethylenic nucleus surface for the positively charged HGEMDA.
The energies of the highest occupied molecular orbitals (EHOMO) generally describe the ability of a compound to give electrons. A high energy value of the HOMO orbital facilitates the tendency of molecules to yield electrons to acceptable species of electrons having unoccupied molecular orbitals with low energy levels. On the other hand, the energies of the lowest unoccupied molecular orbital (ELUMO) are related to the ability of a molecule to accept electrons, a low value of the LUMO energy means that the molecule certainly accepts electrons (Yildiz, 2015). Thus, the adsorption performance of the coating molecule on the metal surface increases when the gap energy (ΔE) decreases (Khaled et al., 2011; El Youssfi et al., 2014).
By analyzing Table 1, we observe that the gap energy values of the neutral, positively charged negative HGEMDA molecule are lower than that of TGEEBA (ΔEgap (HGEMDA) <ΔEgap (TGEEBA)). This indicates that the HGEMDA matrix has a good inhibitory efficacy with respect to TGEEBA. Indeed, we have seen that the positively charged HGEMDA matrix has lower gap energy (1.55 eV) than the negatively charged and neutral charge which is 1.868 eV and 1.874 eV respectively, see Fig. 10.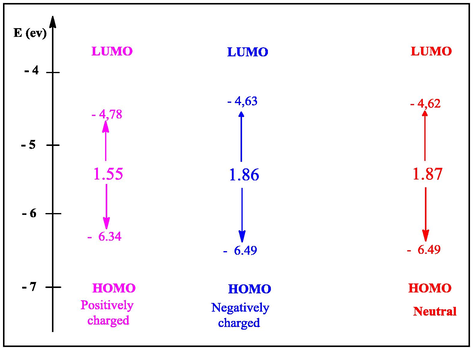
Correlation diagram of the boundary orbitals and the gap energy of the HGEMDA matrix.
The calculated values of hardness (ƞ) and softness (σ) are respectively shown in Table 1. The comparison of the latter shows the two TGEEBA and HGEMDA matrices which can easily react to the metal surface. The HGEMDA matrix has a good chemical reactivity with the surface of the steel that the TGEEBA matrix and this is because of the increase in the values of the softness of the matrix HGEMDA neutral, negatively charged and that positively charged which are respectively equal to 1.067 eV−1, 1.070 eV−1 and 1.287 eV−1. On the other hand, the values of the hardness of the neutral, negatively charged and positively charged HGEMDA matrix are decreased respectively: 0.937 eV, 0.934 eV and 0.777 eV.
Concerning the electrophile index (ω), we have found in Table 1 that the values of the HGEMDA matrix are higher than those of the TGEEBA matrix. For this we have evoked that the neutral, negatively charged and positively charged HGEMDA matrix plays an electron accepting role (very high electrophilic character) which is respectively equal to 16.507 eV, 16.578 eV and 19.935 eV.
As for the charge transfer ΔN for the two matrices TGEEBA and HGEMDA are less than 3.6 eV. This indicates the tendency of a molecule to give electrons to the metal surface (Lukovits et al., 2001; Elmsellem et al., 2015). The inhibitory efficiency increases with the donor capacity of the electrons of the matrices towards the metal surface. In this context of Table 1, we have observed that the transfer of electrons from the neutral, positively charged negative HGEMDA matrix is superior to that of the TGEEBA matrix in the following order ΔN(HGEMDA) > ΔN(TGEEBA). This result indicates that the quality of the film formation is well formed with the HGEMDA matrix.
Consequently, based on the study of all quantum molecular descriptors of epoxy polymers with ethylenic nucleus of two new macromolecular matrices studied TGEEBA and HGEMDA (neutral, negatively charged and positively charged), we have clearly concluded that the electronic effect reacts With the phenomenon of behavior on the epoxy-diamine system.
This result is confirmed by the literature, for a series of macromolecular molecules which differ only in their functional atoms, the efficiency of coating behavior varies inversely with the electronegativity of these functional atoms (Trabanelli, 1987). It therefore increases in the following order: O < N < S < Se < P.
3.2 Electrostatic potential
The electrostatic potential (PES) is defined as the electrostatic interaction energy of a fictitious charge and the determination of this property is very important for the studies of the interactions of the macromolecular matrices. The regions with negative electrostatic potential are favorable to electrophilic attacks while positive regions are more sensitive to nucleophilic attacks (Kosari et al., 2014; Lesar and Milošev, 2009). The electrostatic potential could thus be defined by descriptors which are the Mulliken charge distribution, the molecular electrostatic potential and the molecular electrostatic potential counter.
3.3 Mulliken charge distribution and dipole moment
Fig. 11 shows the distribution of the atomic charges of Mulliken on the atoms of two matrices TGEEBA and HGEMDA (neutral), and thus the dipole moment on these two macromolecular matrices. We have clearly observed in this figure that the atoms of oxygen, nitrogen and some carbon atoms for the two polymer matrices carry negative charges. So, we can say that these atoms are responsible for a nucleophile towards the surface of the steel.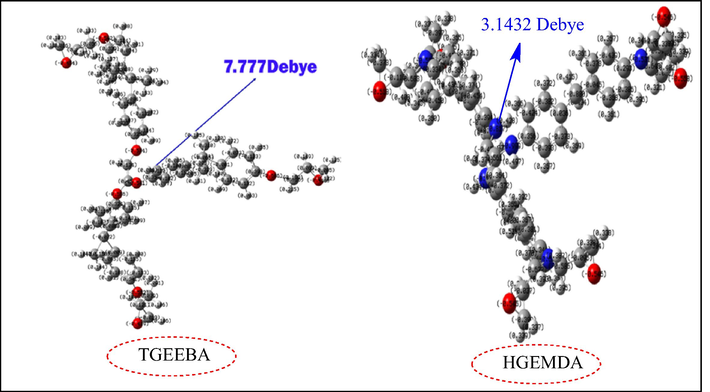
Mulliken charge distribution of two TGEEBA and HGEMDA (neutral) matrices with a vector of the dipole moment.
3.4 Molecular Electrostatic Potential (MESP)
Fig. 12 shows the molecular electrostatic potential of two polymers TGEEBA and HGEMDA and the representations of this potential have been calculated in order to identify the regions of electron density. This strong electron density is presented by a red color and the low electronic density is presented by a blue color. The electron density decreases in the following order: red > orange > yellow > green > blue (Rosline et al., 2014; Panicker et al., 2015; Kaya et al., 2016). The high electron density (yellow to red color) is localized on the oxygen and nitrogen atoms for the two polymer matrices TGEEBA and HGEMDA. The low electron density (green to blue color), on the other hand, is located on a few carbon atoms.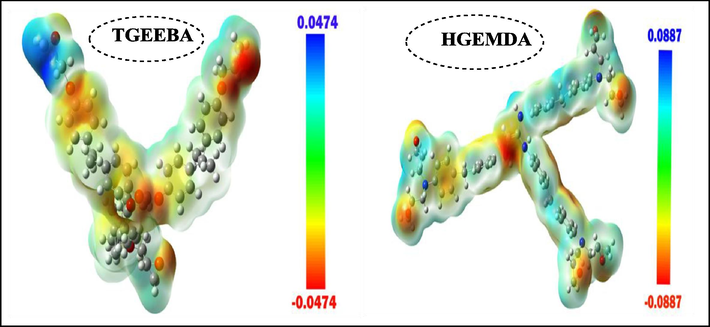
Molecular electrostatic potential of two polymers TGEEBA and HGEMDA.
3.5 Molecular Electrostatic Potential Meter
Fig. 13 shows the account of the molecular electrostatic potential of two polymers TGEEBA and HGEMDA, from which we have noticed that the surface of the electrostatic account for the two matrices TGEEBA and HGEMDA has on the surface of the ethylenic radical and the aromatic nucleains directly bonded to this radical.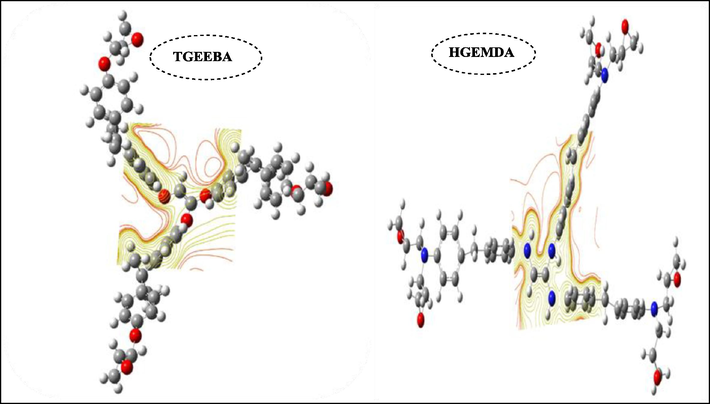
Molecular electrostatic potential of two TGEEBA and HGEMDA polymers.
This study which is carried out with the electrostatic potential for the two new tri- and hexa-functional polyepoxide architectures respectively the triglycidyl ether ethylene of bisphenol A and the hexaglycidyl ethylene of methylene dianiline according to the three descriptors which are the distribution of the charges of Mulliken, the molecular electrostatic potential And the molecular electrostatic potential teller, allowed us to conclude that the effectiveness of the adhesion clearly increases with the number of epoxy functional groups.
3.6 Local reactivity of two macromolecular matrices
The calculation of the Fukui (IF) indices was carried out by using 6–311 G (d, p) basis sets in the gas phase (G), the use of the atomic charge approximation was calculated by analyzing the Natural population (NPA) that was performed in terms of localized electron pair. Electronic density plays an important role in the calculation of the parameters of chemical reactivity. The PNA was performed by using the following equations.
The high value of
measures changes in electron density when the molecule has received an additional electron and
measures changes in electron density when the molecule loses electrons (Eddy et al., 2015; Shahraki et al., 2016). The Fukui indices of two calculated TGEEBA and HGEMDA matrices are shown in Table 2.
Matrices
Atoms
P(N)
P (N + 1)
P (N-1)
f+
f−
TGEEBA
C1
5.98831
5.93265
5.95949
−0.05566
0.02882
C3
5.46244
5.44011
5.45995
−0.02233
0.00249
O4
8.52128
8.52036
8.52879
−0.00092
−0.00751
O5
8.51840
8.50948
8.52977
−0.00892
−0.01137
O6
8.52848
8.52439
8.53752
−0.00409
−0.00904
C7
5.52848
5.69529
5.69700
0.16681
−0.16852
C8
6.26030
6.22331
6.23070
−0.03699
0.02960
C9
6.26382
6.23202
6.22998
−0.03180
0.03384
C10
6.23421
6.19543
6.20642
−0.03878
0.02779
C12
6.23762
6.19942
6.20907
−0.03820
0.02855
C14
6.04438
6.03550
6.05608
−0.00888
−0.01170
C46
5.68572
5.68065
5.67098
−0.00507
0.01474
C47
6.26284
6.21996
6.22841
−0.04288
0.03443
C48
6.28471
6.25300
6.25075
−0.03171
0.03396
C49
6.23208
6.19464
6.20390
−0.03744
0.02818
C51
6.23623
6.20547
6.20781
−0.03076
0.02842
C53
6.05494
6.03361
6.05121
−0.02133
0.00373
C56
5.69119
5.66960
5.68989
−0.02159
0.00130
C57
6.27784
6.24181
6.25390
−0.03603
0.02394
C58
6.25606
6.21007
6.22877
−0.04599
0.02729
C59
6.23442
6.20384
6.20242
−0.03058
0.03200
C61
6.23463
6.19910
6.20373
−0.03553
0.03090
C63
6.04911
6.04369
6.02389
−0.00542
0.02522
HGEMDA
C1
5.72243
5.41081
5.53254
−0.31162
0.18989
C2
6.20135
6.21355
6.47980
0.01220
−0.27845
N3
7.71357
7.64920
7.84073
−0.06437
−0.12716
N4
7.82708
7.49744
7.85014
−0.32964
−0.02306
N5
7.76988
7.84843
7.91766
0.07855
−0.14778
C8
5.35509
5.61569
5.61527
0.26060
−0.26018
C9
6.63329
6.42833
6.43262
−0.20496
0.20067
C10
6.64973
6.43437
6.43724
−0.21536
0.21249
C11
6.58414
6.37484
6.37681
−0.20930
0.20733
C13
6.56026
6.37956
6.38307
−0.18070
0.17719
C15
5.31349
5.90552
5.90550
0.59203
−0.59201
C32
5.10937
5.58508
5.58253
0.47571
−0.47316
C33
6.60470
6.44001
6.44190
−0.16469
0.16280
C34
6.60750
6.44440
6.44639
−0.16310
0.16111
C35
6.24470
6.35716
6.35741
0.11246
−0.11271
C37
6.25052
6.36087
6.36120
0.11035
−0.11068
C39
6.13092
5.95225
5.95225
−0.17867
0.17867
C80
5.85602
5.66716
5.66918
−0.18886
0.18684
C81
6.24956
6.41585
6.45555
0.16629
−0.20599
C82
6.24058
6.42422
6.44862
0.18364
−0.20804
C83
6.24897
6.34473
6.36264
0.09576
−0.11367
C85
6.23624
6.34837
6.39006
0.11213
−0.15382
C87
6.04027
6.03085
6.03379
−0.00942
0.00648
From the analysis of Table 2, we have noticed for the TGEEBA matrix that the highest values of is located on the C (7) atom, while the highest values of are located on the C (48), C (49), C (51), C (57), C (58), C (59), C (61), C (63). These atoms participate in giving electrons to the metal surface.
For the hexa-functional matrix HGEMDA, we found that the C (8), C (15), C (32), C (35), C (C), C (13), C (33), C (34), C (39) and C (80) are involved in the electrophilic attack. This confirms the previous results obtained by the electrostatic potential. The application of molecular dynamics would constitute a new confirmatory approach to this molecular modeling.
3.7 Monte Carlo simulations of two epoxy polymers
Monte Carlo simulations were performed to further study the adsorption behavior of polymers on the Fe (1 1 0) surface. Thus, MC simulation reasonably predicts the most favorable configuration of the adsorbed polymer on the low adsorption energy metal surface. In this context, the most stable adsorption configurations of the polymers studied on the Fe (1 1 0) surface using Monte Carlo simulations are presented in Figs. 14 and 15. From the careful observation of these figures, it can be said that the polymers studied adsorb on the carbon steel surface in a parallel mode under all circumstances, the adsorbed polymers also cover a very important part of the steel surface, indicating that the recovery capability for the corrosion of the carbon steel studied is reasonable.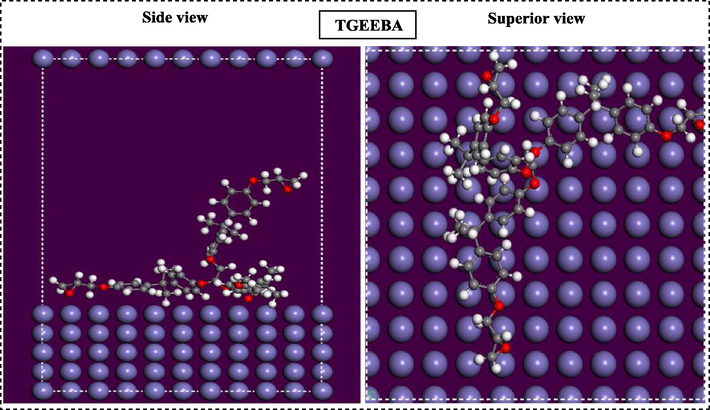
Most stable adsorption configuration of low energy of trifunctional polymer TGEEBA.
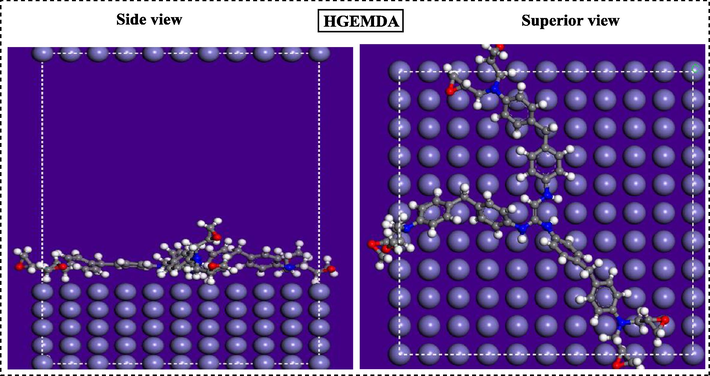
Most stable adsorption configuration of low energy of hexafunctional polymer HGEMDA.
The different values of the energies obtained from the Monte Carlo simulation, namely the total energy, the adsorption energy, the rigid adsorption energy and the deformation energy are collated in Table 3. In addition, (dEad/dE (polymer)) is the energy required to remove an adsorbate from the metal surface, a low deformation energy due to adsorbate relaxation on the surface Fe (1 1 0) is the most important. The high absolute value of the adsorption energy reflects a strong adsorption behavior. Indeed, the matrix HGEMDA has a low value of the adsorption energy than that of the TGEEBA, indicating that this polymer has a high adsorption capacity on the metal surface.
Polymers
Total Energy
Adsorption Energy
Rigid adsorption energy
deformation energy
dEad/dE (polymer)
Fe(1 1 0)/HGEMDA
−213.34
−604.70
−152.28
−452.42
−604.70
Fe(1 1 0)/TGEEBA
−331.46
−291.77
−239.57
−52.20
−291.77
In parallel with the above, the order of adsorption energy is confirmed with the protection efficiency obtained from the experimental results and density functional theory (DFT).
4 Conclusion
The elaboration of two new architectures with polyepoxide ethylenic nucleus and tri- and hexa-functionality, respectively; Triglycidyl ether of bisphenol A (TGEEBA) and hexaglycidyl ethylene of methylene dianiline (HGEMDA). The prediction of the quantum molecular descriptors is based on the HOMO and LUMO energies through the Gaussian software 03 which are E (HOMO), E (LUMO), (I), (A), ΔE, σ, ƞ, χ, and ΔN. The obtained results show that the efficiency of the hexafunctional matrix is more dominant than that of the trifunctional macromolecular matrix on the one hand while on the other hand, to confirm the previous theoretical study, we first extended our study to the evaluation of a theoretical descriptor. The latter is the electrostatic potential which is based on the distribution of Mulliken charges, electrostatic potential and molecular electrostatic potential. These two new studies confirm that the hexafunctional matrix has a more efficient coating than that of the trifunctional matrix. Indeed, we conclude that the matrix HGEMDA has a low value of the adsorption energy than that of the TGEEBA, indicating that this polymer has a high adsorption capacity on the metal surface. Finally, the simulation of the Monte Carlo confirms the method of the DFT.
References
- Experimental and quantum chemical evaluation of Schiff bases of isatin as a new and green corrosion inhibitors for mild steel in 20% H2SO4. J. Aiwa. Inst. Chem. Eng.. 2015;54:145-154.
- [Google Scholar]
- Density functional calculations of molecular bond energies. J. Chem. Phys.. 1986;84(8):4524-4529.
- [Google Scholar]
- Synthesis, structural, viscosimetric, and rheological Study, of a new trifunctional phosphorus epoxyde prepolymer, tri-glycidyl ether tri-mercaptoethanol of phosphore (TGETMEP) Medit. J. Chem.. 2016;6:665-673.
- [Google Scholar]
- Prediction of Polymer Propertie. New York: Marcel Dekker; 1996.
- Application of a mathematical model to the study of the glass transition temperature in polymer electrolyte precursor systems. Polym. Bull.. 1997;38(3):353-357.
- [Google Scholar]
- Theoretical and experimental studies on the corrosion inhibition potentials of some purines for aluminum in 0.1 M HCl. J. Adv. Res.. 2015;6:203-217.
- [Google Scholar]
- Corrosion protection of mild steel by 6-chloroquinoxaline-2,3(1H,4H)-dione as a new inhibitor in hydrochloric acid solution. J. Mater. Environ. Sci.. 2016;7(11):4311-4323.
- [Google Scholar]
- Theoretical prediction and experimental study of 5-methyl-1H-pyrazole-3- carbohydrazide as a novel corrosion inhibitor for mild steel in 1.0 M HCl. Der Pharma Chemica. 2015;7(10):237-245.
- [Google Scholar]
- Der Pharma Chemica. 2014;7:284-291.
- Prediction of indices of refraction and glass transition temperatures of linear polymers by using graph theoretical indices. J. Phys. Chem. B. 2002;106(6):1501-1507.
- [Google Scholar]
- Prediction of the glass transition temperatures for epoxy resins and blends using group interaction modeling. J. Polym.. 2001;42(13):5717-5725.
- [Google Scholar]
- Theoretical studies of three triazole derivatives as corrosion inhibitors for mild steel in acidic medium. Corros. Sci.. 2014;87:366-375.
- [Google Scholar]
- Viscosimetric and rheological studies of a new trifunctional epoxy pre-polymer with noyan ethylene: triglycidyl Ether of Ethylene of Bisphenol A (TGEEBA) J. Mater. Environ. Sci.. 2017;8:603-610.
- [Google Scholar]
- Synthesis, characterization, viscosimetric study and formulation of a new nanofunctional polymer’s nanocomposite: nanoglycidyl trihydrazine 4,4,4-tripropoxy tribisphenol A of ethylene. Inter. J. Sci. Eng. Res.. 2017;8:1181-1188.
- [Google Scholar]
- Application of a new tri-functional epoxy prepolymer, triglycidyl ethylene ether of bisphenol A, IN THE COATING OF E24 STEEL IN 3.5 % NaCl. J. Chem. Tech. Metal.. 2017;52(3):431-438.
- [Google Scholar]
- Synthesis and characterization of a new octafunctional epoxy resin (Octaglicydil tetra p-aminophenol of bisphenol A (Bis para phosphoric ester)). Viscosimetric study. Mor. J. Chem.. 2015;3:791-797.
- [Google Scholar]
- Theoretical study of the impact of quantum chemistry parameters on the behavior all effects of the coating of a new epoxy prepolymer. Mor. J. Chem.. 2018;6:35-42.
- [Google Scholar]
- Theoretical and experimental studies of a new tri-functional polyepoxide: triglycidyl Ethylene Ether of Bisphenol A (TGEEBA). Optimisation of parameters by the experimental design and the formulation of a nanocomposite. Arab. J. Chem. Environ. Res.. 2016;3:35-50.
- [Google Scholar]
- Neural network prediction of glass-transition temperatures from monomer structure. J. Chem. Soc. Faraday Trans.. 1995;91:2491-2496.
- [Google Scholar]
- Prediction of glass transition temperatures from monomer and repeat unit structure using computational neural networks. J. Chem. Inform. Comput. Sci.. 2002;42:232-240.
- [Google Scholar]
- Quantum chemical and molecular dynamics simulation studies on inhibition performances of some thiazole and thiadiazole derivatives against corrosion of iron. J. Mol. Liq.. 2016;219:497-504.
- [Google Scholar]
- Molecular level investigation of the interaction of cerium dioxide layer on steel substrate used in refrigerating systems. J. Mater. Environ. Sci.. 2011;2(2):166-173.
- [Google Scholar]
- Electrochemical and quantum chemical assessment of two organic compounds from pyridine derivatives as corrosion inhibitors for mild steel in HCl solution under stagnant condition and hydrodynamic flow. Corros. Sci.. 2014;78:138-150.
- [Google Scholar]
- Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785-789.
- [Google Scholar]
- Density functional study of the corrosion inhibition properties of 1,2,4-triazole and its amino derivatives. Chem. Phys. Lett.. 2009;483:198-203.
- [Google Scholar]
- Corrosion inhibitors—correlation between electronic structure and efficiency. Corrosion. 2001;57(1):3-8.
- [Google Scholar]
- Max Planck Institute for Bioinorganic Chemistry. Germany: Mulheim & Ruhr; 2012.
- FT-IR, HOMO-LUMO, NBO, MEP analysis and molecular docking study of 3-Methyl-4-{(E)-[4-(methylsulfanyl)-benzylidene]amino}1H-1,2,4-triazole-5(4H)-thione. Spectrochim. Acta. A. Mol. Biomol. Spectrosc.. 2015;151:198-207.
- [Google Scholar]
- Absolute hardness: companion parameter to absolute electronegativity. J. Am. Chem. Soc.. 1983;105(26):7512-7516.
- [Google Scholar]
- FT-IR, FT-Raman, molecular structure, first order hyperpolarizability, HOMO and LUMO analysis, MEP and NBO analysis of 3-(adamantan-1-yl)-4-(prop-2-en-1-yl)-1H-1,2,4-triazole-5(4H)-thione, a potential bioactive agent. Spectrochim. Acta. A. Mol. Biomol. Spectrosc.. 2014;132:295-304.
- [Google Scholar]
- Molecular dynamics and density functional theory study on corrosion inhibitory action of three substituted pyrazine derivatives on steel surface. Can. Chem. Trans.. 2014;2(4):489-503.
- [Google Scholar]
- Evaluating electronic structure of quinazolinone and pyrimidinone molecules for its corrosion inhibition effectiveness on target specific mild steel in the acidic medium: a combined DFT and MD simulation study. J. Mol. Liq.. 2016;224:629-638.
- [Google Scholar]
- Theoretical studies on the corrosion inhibition performance of three amine derivatives on carbon steel: molecular dynamics simulation and density functional theory approaches. J. Taiwam Inst. Chem. Eng.. 2016;62:313-321.
- [Google Scholar]
- Molecular modelling of polymers, 3*. Prediction of glass transition temperatures of poly(acrylic acid), poly(methacrylic acid) and polyacrylamide derivatives. Macromol. Theor. Simul.. 1996;5(3):467-475.
- [Google Scholar]
- Adsorption and corrosion inhibition behavior of mild steel by one derivative of benzoic−triazole in acidic solution. Ind. Eng. Chem. Res.. 2010;49(6):2593-2599.
- [Google Scholar]
- Corrosion inhibitors. In: Mansfeld F., ed. Corrosion Mechanisms. New York: Marcel Dekker; 1987.
- [Google Scholar]
- Simulation study of the glass transition temperature in poly(methyl methacrylate) Phys. Rev. E. 2002;65:21805.
- [Google Scholar]
- Computational neural networks and the rational design of polymeric materials: the next generation polycarbonates. Comput. Theor. Polym. Sci.. 1998;8(3–4):311-321.
- [Google Scholar]
- A relationship between the glass transition temperature (Tg) and fractional conversion for thermosetting systems. J. Appl. Polym. Sci.. 1997;64(1):3-14.
- [Google Scholar]
- An electrochemical and theoretical evaluation of 4,6-diamino-2-pyrimidinethiol as a corrosion inhibitor for mild steel in HCl solutions. Corros. Sci.. 2015;90:544-553.
- [Google Scholar]
- Synthesis and physico chemical study of a new hexa and tetra functional epoxy materials based on bis -para-terephthalylidene phosphoric ester. J. Mater. Environ. Sci.. 2010;1:213-218.
- [Google Scholar]




