

Translate this page into:
In silico study for evaluating the binding mode and interaction of 1, 2, 4-triazole and its derivatives as potent inhibitors against Lipoate protein B (LipB)
⁎Corresponding author. shola4343@gmail.com (Shola Elijah Adeniji)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Tuberculosis (TB) is an infectious disease caused by bacterium specie known as Mycobacterium tuberculosis. Emergence of multi-drug resistant strains of M. tuberculosis led to the development of new and more potent anti-tuberculosis agents. A novel series of 1, 2, 4-triazole derivatives have been reported as better anti-tubercular agents. Thus, Lipoate biosynthesis protein B (LipB) was selected as a potential drug target and docked with the inhibitors to evaluate the binding mode and interaction. The Molecular docking analysis showed that nearly all the compounds bind strongly to active sites of the target with binding affinities ranging from (−4.1 to −17.9 kcal/mol) which correlates with their activities. Ligands (compound 16 and 34) have the best binding affinity of (−15.8 and −17.9 kcal/mol) which formed hydrophobic interaction and hydrogen bond with amino acid residues of M. tuberculosis Lipoate protein B (LipB). This research has shown that the binding affinity of these compounds were found to be better than the recommended anti-mycobacterium drugs; isoniazid (−14.6 kcal/mol) and ethambutol (−5.8 kcal/mol). This study provides a valuable approach for designing and synthesizing more potent anti-mycobacterium tuberculosis derivatives.
Keywords
Binding affinity
1, 2, 4-triazole
Molecular docking
M. tuberculosis
LipB

1 Introduction
Tuberculosis still remains a major challenge to mankind caused by Mycobacterium tuberculosis. There are drugs like ethambutol, isoniazid and rifampicin available for curing for tuberculosis. The increasing problem of Multi-Drug Resistant (MDR) tuberculosis has focused the attention of researchers toward the development of novel drugs that are not only shortening the prolonged therapy but also active against disease. (Lamichhane et al., 2011; Maste et al., 2011).
In developing and designing of novel anti-tubercular drugs, it is very important to think about which receptor in the tubercle bacillus is a good drug target. There are many enzymes that partake in the pathogenicity and metabolic process like the growth of the bacterium and one among them is Lipoate biosynthesis protein B (LipB). LipB is an enzyme that participates in lipoylation; it catalyzes the transfer of endogenous octanoic acid to lipoyl domains by forming thioester bond to the 4-phosphopanthetheine cofactor of the acyl carrier protein (ACP). Lipoyl synthase (Lip A) then converts octanoyl derivatives into lipoyl derivatives. Thus it acts as the essential protein involved in activating the bacterium’s metabolic activities (Cade et al., 2010).
Computational approach aids to evaluate the binding affinity and interaction between a ligand and the receptor which help to prioritize drug for screening experimental approach. Protein-ligand docking is a computational method developed to understand the binding mode and interpret the preferred orientation between large molecules (receptor) and small molecule (ligand) in order to form a stable complex. This technique plays a vital role in computer aided drug design (Kitchen et al., 2004). Molecular docking investigations were carried out with the aim of understanding the binding mode and interaction of the 1, 2, 4-triazole derivatives into the active site of LipB of M. tuberculosis.
2 Materials and method
2.1 Optimization
The chemical structures of the molecules were drawn with Chemdraw ultra Version 12.0. Each molecule was first pre-optimized with the molecular mechanics (MMFF) and further re-optimize with Density functional theory (DFT) utilizing the B3LYP and 6-31G* basis set (Becke, 1993; Lee et al., 1988). The Spartan files of all the optimized molecules were then saved in PDB file format which is the recommended input format in Discovery studio version 1.4.5 and Discovery Studio Visualizer software.
2.2 Docking procedure
The molecular docking studies were carried between 1, 2, 4-triazole derivatives and M. tuberculosis target site (LipB). The molecular structures 1, 2, 4-triazole derivatives were presented Table 1. These compounds together with their biological activity were obtained from the literature (Sarkar et al., 2016). While the crystal structure of (LipB) was obtained from the Protein Data Bank with code 1W66. All bound substances (ligands and cofactors) and solvent molecules associated with the receptor were removed. Some of the active sites are GLN, ALA, ASP, PHE, CYS, ASN, SER etc. The prepared receptor and ligand were shown in Fig. 1. The prepared ligands were docked with the prepared structure of LipB using Autodock Vina incorporated in Pyrx software. The docked results were then visualized and analyzed using Discovery Studio Visualizer software (Adeniji et al., 2020).
S/N
Molecule
Activity
(% inhibition)
1
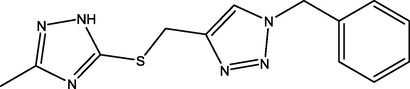
03
2
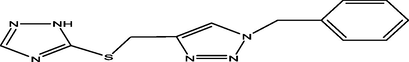
10
3
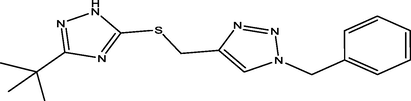
03
4
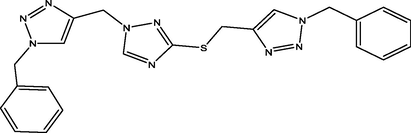
11
5
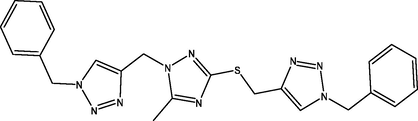
08
6
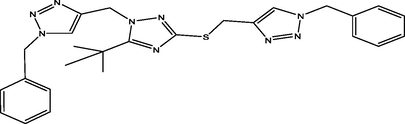
08
7
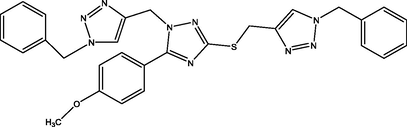
01
8
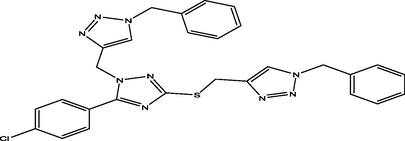
21
9
08
10
85
11
10
12
13
13
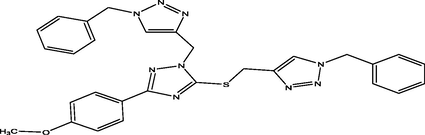
86
14
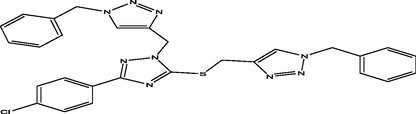
31
15
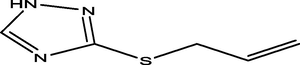
51
16
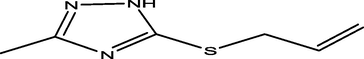
98
17
57
18
25
19
94
20
98
21
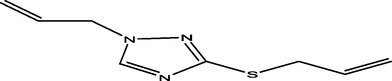
58
22
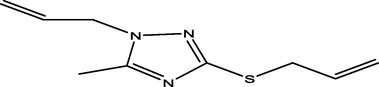
25
23
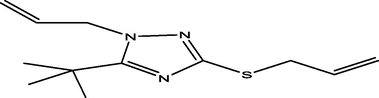
45
24
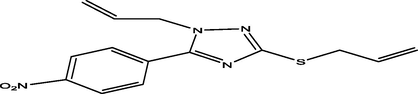
15
25
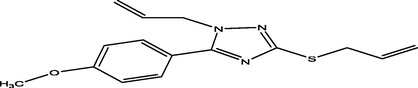
89
26
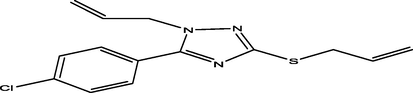
94
27
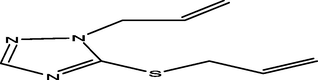
65
28
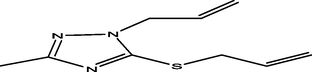
22
29
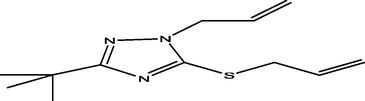
30
30
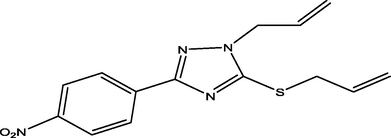
40
31
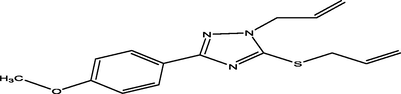
88
32
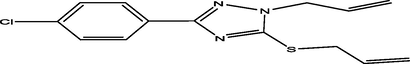
26
33
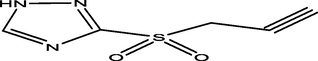
42
34
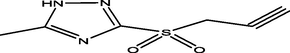
98
35
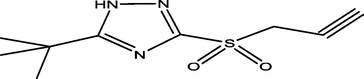
76
36
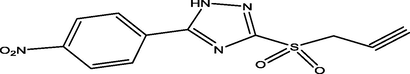
96
37
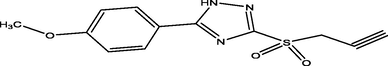
88
38
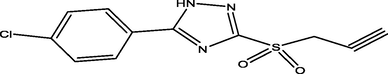
88
39
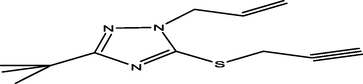
76
40
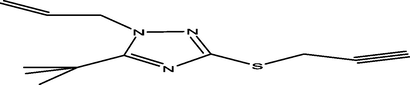
96
41
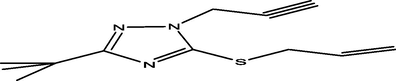
98
42
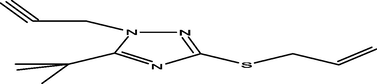
98
43
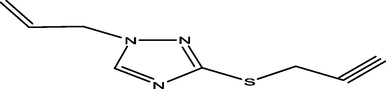
98
44
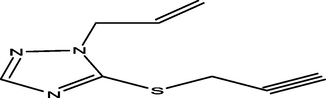
40
45
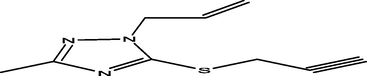
98
46
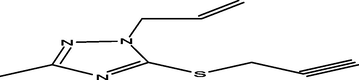
98
47
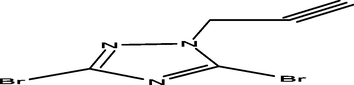
97
48
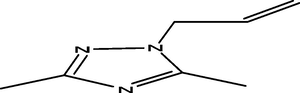
12
49
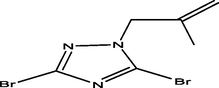
09
50
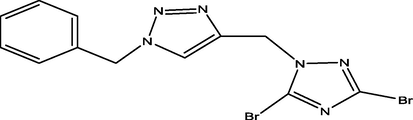
02
Prepared structure of LipB.
3 Results and discussion
Molecular docking studies were carried out in order to elucidate the interaction and the binding mode between the target (LipB) and 1, 2, 4-triazole derivatives as potent anti-mycobacterium tuberculosis. The docking results clearly show that the binding affinities of these ligands correlate with their activity values. The binding affinity values for all the compounds range from (−4.1 and 17.9 kcal/mol) as reported in Table 2. Ligands (compound 16 and 34) have higher binding affinities which ranges from (−15.8 to 17.9 kcal/mol) which were greater than the binding affinity of recommended drugs; isoniazid (−14.6 kcal/mol) and ethambutol (−5.8 kcal/mol). Ligands (compound 16 and 34) with best binding affinities were visualized and analyzed using Discovery Studio Visualizer. The 2D and 3D interaction of ligand 16 and 34 as well as recommended anti-tubercular drugs (ethambutol and isoniazid) with LipB target site were shown in Figs. 2 and 3.
Ligand
Binding Affinity (BA)
kcal/molHydrogen bond
Hydrophobic interaction
Amino acid
Bond length (Ao)
Amino acid
1
−4.2
–
–
PHE243, ALA167
2
−6.3
VAL112
1.3452
ALA203, PHE130, VAL78
3
−4.4
–
–
PHE128, VAL78, PRO232, VAL128, SER237
4
−6.5
THR87
1.4234
ALA237, TRP123, LEU154, VAL228
5
−6.1
THR78
1.2433
ALA167, TRP122, LEU184, VAL228, VAL73
6
−6.2
ALA1 23
1.2233
PHE248, VAL228, CYS143, LEU176
7
−4.1
–
–
TRP182, ALA167, VAL78, SER247, CYS145
8
−7.6
ALA167
2.4332
CYS221, TRP182, ALA212, PRO165
9
−6.3
GLN385
1.3443
ALA143, TRP182, PHE168
10
−12.4
THR77
GLN3852.4554
2.4332LEU164, VAL78, VAL228, ALA236
11
−6.2
ASN74
1.3454
PRO134, VAL78, LA167, ALA233
12
−6.6
GLN385
1.6445
VAL83, VAL83, LEU76, TRP182
13
−12.8
LEU103
TRP1822.3421
3.0328ALA233, PRO346, ALA167
14
−7.9
ASN74
2.7656
ALA167, LEU164, VAL83,
15
−8.7
PHE164
ASP282.1836
2.2223LEU164, VAL78, VAL82, PRO285
16
−15.8
ASP110
PHE109
ALA1112.3503
2.1532
2.6856TYR113, PRO112
17
−8.8
GLN385
CYS3452.7332
2.4333VAL78, ALA233, TRP182, VAL78
18
−7.6
GLN385
2.5433
PRO285, PHE168, ALA167, VAL83, PRO285, VAL83
19
−14.7
VAL78
ALA233
LEU762.1322
2.4876
2.4517SER237, THR238, PHE168, PRO285, VAL78, ALA167,
20
−14.8
GLN385
ARG386
GLN1052.5684
2.4569
2.0487PRO94, PRO34, PHE93, VAL178, PRO169, PHE241, PHE338, CYS345
21
−8.7
ASN78
ASP2323.0175
2.2831LEU207, VAL228, LEU73, VAL78, PRO245
22
−7.7
THR77
2.4532
PHE168, TRP182, TRP182, PHE168, VAL78, ALA167
23
−8.2
GLN385
SER2372.1265
2.2453PRO285, PHE338, CYS345, VAL78, ALA233
24
−6.9
TRP182
1.7232
VAL82, PRO285, VAL78, VAL78, ALA167, PRO285
25
−14.2
ASP282
LYS136A
GLN3852.1238
2.1433
2.2334LEU103, VAL78, TRP182, ALA167, PRO285
26
−14.9
GLN105
ALA167
VAL822.2339
2.2344
2.5753LYS173, ALA128, PHE168, TRP182, PHE230, ALA111, PRO112, VAL82, VAL78
27
−9.3
ASP78
GLN3853.3648
2.4850PRO346, ALA167, PHE168, TRP182, CYS345, ALA233
28
−7.6
VAL77
2.4322
TRP182, ALA167, TRP182, PRO285, VAL27, PRO34
29
−7.8
ASN74
3.4567
VAL99, PHE280, VAL142
30
−8.0
GLN385
LEU1032.17739
2.2281VAL78, ALA233, LEU161, PHE168, TRP18
31
−14.4
GLN385
CYS1702.0343
2.1732PHE215, LEU207, MET66, VAL78, ALA147, PRO94
32
−7.9
VAL95
2.6433
LEU217, TYR113, PRO112, VAL78
33
−8.3
GLN105
ARG722.5433
2.1843ALA137, VAL122, TRP182, PHE220
34
−17.9
THR77
GLN385
ALA167
GLN385
ALA1872.1123
2.6234
2.6012
2.1922
2.6302PHE168, VAL78
35
−10.7
THR77
ALA167
GLN3852.1423
2.3432
2.134GLY232, VAL228, PHE168, TRP182, LYS175, ALA233
36
−14.7
PHE164
CYS134
GLY2322.2211
2.211
2.3732PHE168, TRP182, PRO169, LYS136, VAL78, ALA167,
37
−14.3
GLU98
PRO134
ALA1672.0629
2.3934
2.5443LEU103, ALA167, VAL78, ALA233, PRO285, PHE168
38
−14.1
PRO94
ARG97
VAL952.4532
2.1023
2.5434TRP182, TRP182, PRO285, PHE168, VAL142,
39
−10.8
VAL78
ASN74
GLN3852.3647
2.0362
2.0232VAL228, LEU164, VAL78, ALA233, PRO285, ALA137, ALA233,
40
−14.8
ALA167
GLN385
LEU1372.2475
2.2345
2.5434CYS254, PHE168, TRP182, VAL78, ALA167, VAL142, LEU103
41
−15.3
ALA233
PRO94
GLN852.3091
2.2823
2.211GLY232, VAL228, PHE168, LEU164, VAL228
42
−15.1
GLN385
CYS234
ARG3862.1563
2.2793
2.2584VAL228, ALA233,
43
−15.6
PRO94
HIS343
ALA2333.0502
2.1334
1.2445CYS345, PHE 168, ALA176, GLN 322, TRP182, ARG72, GLN385, VAL78
44
−7.3
ALA167
GLN3853.7443
1.3444ALA167, PHE280, ALA233, THR77
45
−14.7
GLN385
ALA167
VAL772.16131
2.3440
1.4343ALA167, ARG386, ALA281, LEU164, VAL228, TRP182
46
−14.9
LEU134
GLN345
ARG1452.3441
2.3234
1.2322VAL178, PRO169, LEU164, VAL228,
47
−13.8
ARG165
GLN385
ARG3861.99395
2.3433
2.4551ALA167, PHE185, VAL228, CYS134, ASN74
48
−7.0
THR65
1.43511
CYS170, ALA233, GLN385
49
−6.3
GLN385
1.322
ARG165, GLN385, CYS234, VAL167, GLN385
50
−4.2
–
–
LEU103, GLY96, PHE205, ARG101, LEU207
Ethambutol
−5.8
ALA337
2.59739
–
Isoniazid
−14.6
SER279
ALA337
ALA3372.29943
2.52954
2.24657PHE338
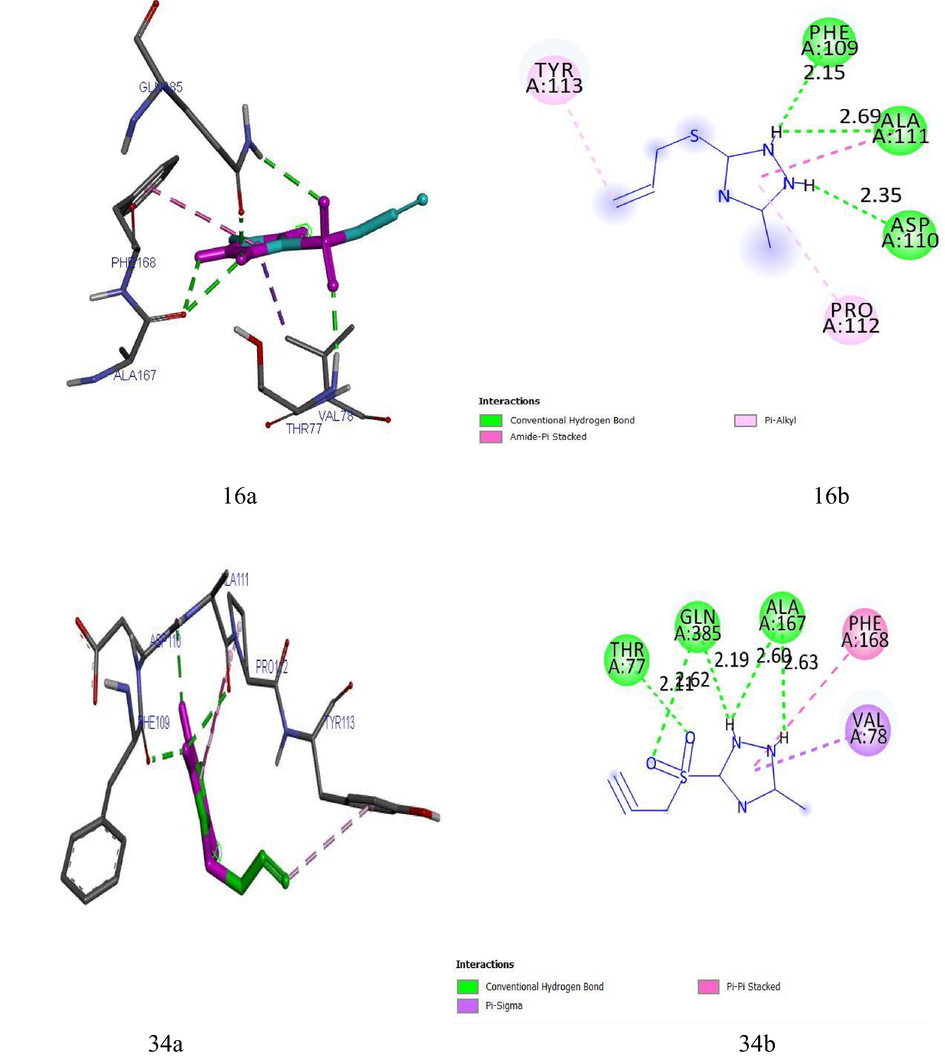
(16a) and (16b) show the 3D and 2D interactions between LipB and Ligand 16. (34a) and (34b) show the 3D and 2D interactions between LipB and Ligand 34.
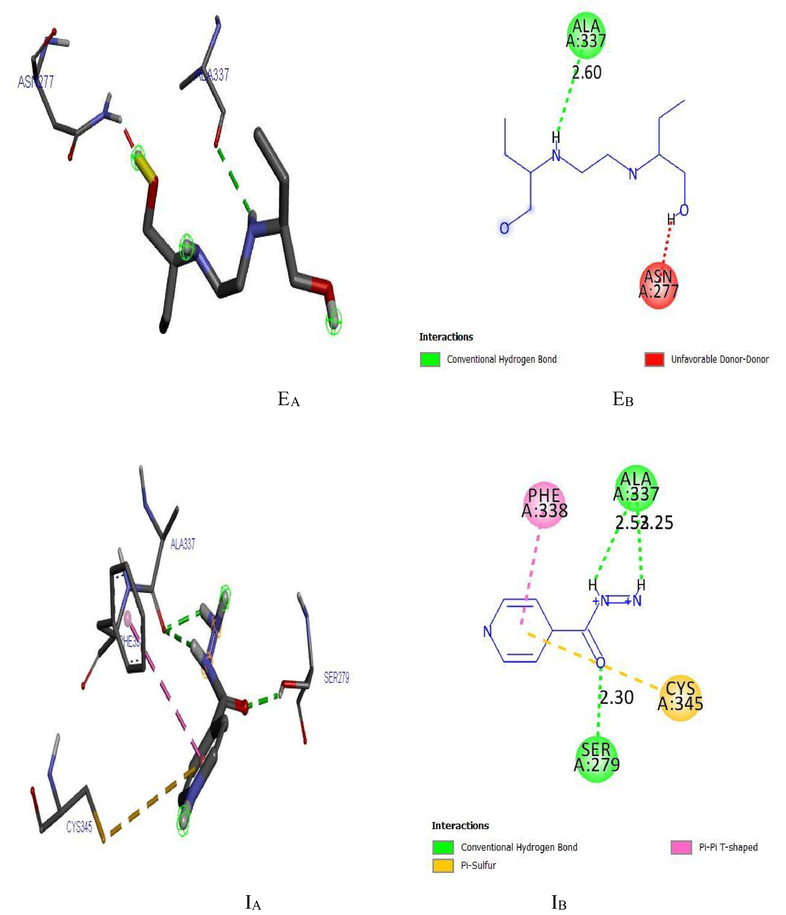
(EA) and (EB) show the 3D and 2D interactions between LipB and Ethambutol. (IA) and (IB) show the 3D and 2D interactions between LipB and Isoniazid.
Ligand 16 formed three hydrogen bonds (2.3503, 2.1532 and 2.6856 A) with TYR113 and PRO112 of the target. Hydrophobic interaction is a bond formed between the ligand and the binding pocket of the target site (receptor). It adhere the ligand to the surface of target site as shown in Figs. 4a and 4b. Ligand 16 formed hydrophobic bond with ASP110, PHE109 and ALA111 of the target site. Ligand 34 formed five hydrogen bonds (2.1123, 2.6234, 2.6012, 2.1922 and 2.6302 A) with THR77, GLN385, ALA167, GLN385 and ALA187 of the target while hydrophobic interactions were observed PHE168 and VAL78. The recommended drugs; Isoniazid formed three hydrogen bonds (2.53, 2.25 and 2.30 A) with ALA337, ALA337 and SER273 while hydrophobic bonds were observed with PHE338 and CYS345 while ethambutol formed only one hydrogen bond (2.60°A) with ALA337 with the target site but no hydrophobic interaction which accounts for its low binding affinity.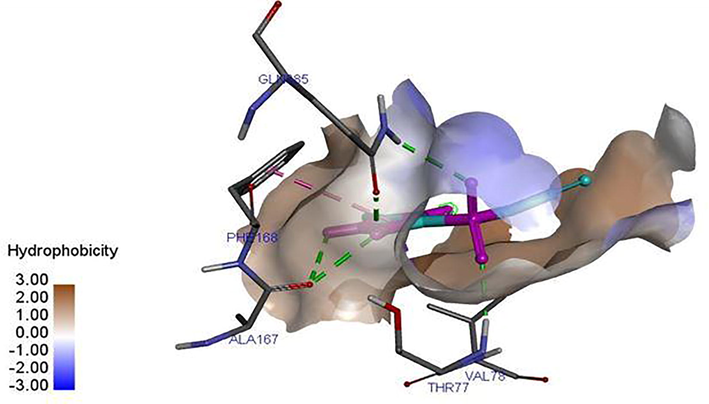
Hydrophobic interaction between the ligand 16 and M. tuberculosis target (LipB).
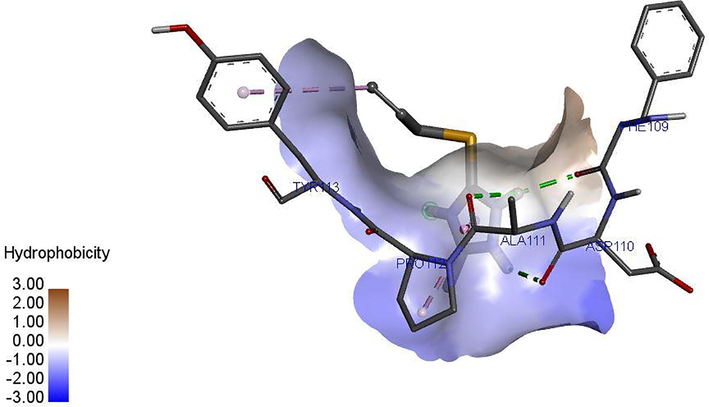
Hydrophobic interaction between the ligand 34 and M. tuberculosis target (LipB).
Ligand 16 formed a total of three hydrogen bonds with target site of LipB. The N-H group triazolidine of the ligand acts as hydrogen donor and formed three hydrogen bonds with PHE109, ALA11 and ASP110 of the target. Ligand 34 formed a total of five hydrogen bonds with target site of LipB. The N-H group triazolidine of the ligand also acts as hydrogen donor and formed three hydrogen bonds with GLN385, ALA167, and ALA167 of the target. The S⚌O of the ligand acts as hydrogen acceptor and formed two hydrogen bonds with ASN79 and GLN 385 of the target. The hydrogen bond formation alongside with the hydrophobic interaction provides an evidence that ligand 16 and 34 of the inhibitor compounds are potent against LipB receptor. Elucidations of hydrogen donor and hydrogen acceptor region were shown in Figs. 4c and 4d.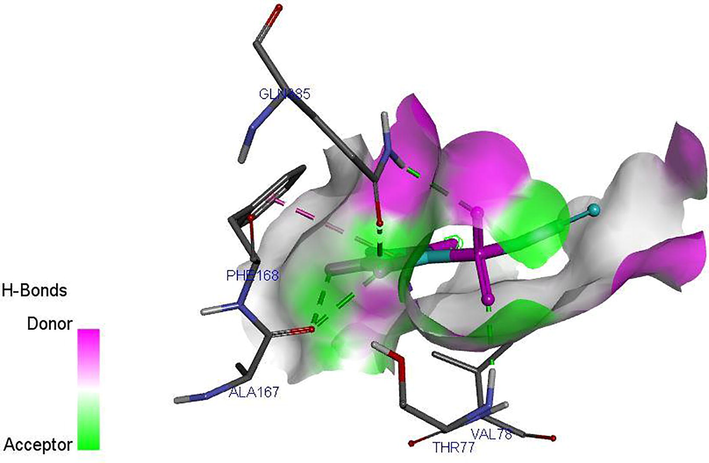
H-bond interaction between the ligand 16 and M. tuberculosis target (LipB).
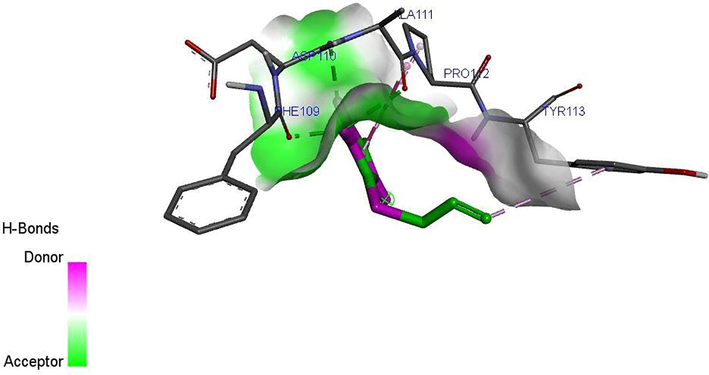
H-bond interaction between the ligand 34 and M. tuberculosis target (LipB).
4 Conclusion
Analogue of 1, 2, 4-triazole derivatives were evaluated against Mycobacterium tuberculosis target (LipB). The binding affinities of these compounds correlate with their biological activities. Ligands (compound 16 and 34) were found to have the most promising binding affinity values of (−15.8 and 17.9 kcal/mol). In conclusion, this study showed that compound 16 and 34 of 1, 2, 4-triazole derivatives could serve as better anti-tuberculosis drug and need further in vitro investigations to confirm their actual therapeutic potential efficacy and drug ability towards the disease.
References
- Computational modeling of 4-Phenoxynicotinamide and 4-Phenoxypyrimidine-5-carboxamide derivatives as potent anti-diabetic agent against TGR5 receptor. J. King Saud Univ. -Sci.. 2020;32:102-115.
- [Google Scholar]
- Becke’s three parameter hybrid method using the LYP correlation functional. J. Chem. Phys.. 1993;98:5648-5652.
- [Google Scholar]
- Isoniazid-resistance conferring mutations in Mycobacterium tuberculosis KatG: catalase, peroxidase, and INH-NADH adduct formation activities. Protein Sci.. 2010;19:458-474.
- [Google Scholar]
- Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discov.. 2004;3:935-949.
- [Google Scholar]
- Essential metabolites of Mycobacterium tuberculosis and their mimics. MBio. 2011;2 e00301–10
- [Google Scholar]
- Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785.
- [Google Scholar]
- Triazolone and their derivatives for anti-tubercular activities. Asian J. Res. Chem.. 2011;4:1050-1054.
- [Google Scholar]
- Sarkar, D., Deshpande, S.R., Maybhate, S.P., Likhite, A.P., Sarkar, S., Khan, A., Chaudhary, P.M., Chavan, S.R., 2016. 1, 2, 4-triazole derivatives and their anti-microbial activity. United states patent (US 9376402B2).




