

Translate this page into:
In silico prediction of deleterious single nucleotide polymorphism in human AKR1C3 gene and identification of potent inhibitors using molecular docking approach
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Various computational methods are employed to analyze the various non-synonymous single nucleotide polymorphisms (nsSNPs) in the AKR1C3 gene, that is shown with the protein based approaches along with suitable inhibitors.
Abstract
Objective
The Aldo-keto reductase family consists of a number of enzymes which are essential to the catalysis of redox transformation and which are also involved in intermediate metabolism and detoxification. Several studies have been reported that the catalytic-dependent function of AKR1C family members isoforms and their essential roles in various cancer types including prostate cancer and play a key role in drug resistance and drug detoxification. The aim of the current study was to predict and analyze the deleterious single nucleotide polymorphism (SNP) that is highly associated with prostate cancer. In addition, to find the potent bioactive compounds as effective inhibitors against prostate cancer.
Methods
Various computational methods are employed to analyze the various non-synonymous single nucleotide polymorphisms (nsSNPs) in the AKR1C3 gene.
Results
A total of 18,594 SNPs data set of deleterious and non-coding synonymous were retrieved from the dbSNP database followed by various computational SNP prediction tools were performed to find the most deleterious nsSNP. A total of eight high-risk nsSNPs were predicted and most of the residue is present in the structural and functional conserved domain, hence, both wild type and mutant forms of AKR1C3 were selected for structural analysis. Besides, molecular docking, ADME, and Prime MM/GBSA calculations were also performed with plant derived bioactive compounds with AKR1C3 receptors. The results of the study depicted that the rs62621365 and its possible mutation A258C was considered as the most deleterious nsSNP and plant compounds such as Ginkgetin and Withaferin A shows best binding affinity with both wild type and mutant from of AKR1C3.
Conclusion
The overall results depicted that nsSNPs may be considered for risk assessment against prostate cancer and for cure, the suggested plant derived bioactive compounds may act as potent inhibitors.
Keywords
AKR1C3
nsSNP
SIFT
PolyPhen
Molecular docking
ADME
MM/GBSA

1 Introduction
Aldo-keto reductase family 1 C3 (AKR1C3) is one of the essential members of the Aldo-keto reductase family that play a crucial role in oxidoreductase with a number of substrates involved in the production of extra-testicular androgens (Karunasinghe et al., 2017). It highly expressed in major organs like liver, kidney, brain, placenta, and testis, though it abundantly expressed in the liver hence it significantly involved in steroid hormone metabolism (AKR1C1-AKR1C3) and metabolism of bile salts (AKR1C4) and also it plays an essential role in detoxification of drugs. Among the 4 members, except AKR1C4, these enzymes may be a crucial factor of tobacco-induced cancers including prostate and breast cancer types (Chen and Zhang, 2012). A number of environmental factors and genetic variants have significantly influenced the activity of AKR1C3 (Liu et al., 2008). Mainly the genetic variants are essential to produce variable testosterone expression levels and for the activation of androgen receptor (AR) (Davey and Grossmann, 2016). Prostate cancer is the second most cancer type commonly occurring in men and fourth-most cancer commonly occurring overall cancer types (Rawla, 2019). The highest rate of prostate cancer was recorded in countries including France, Ireland, Norway, and Sweden. American chemical society reported that about 191, 930 new cases and 33,330 deaths from prostate cancer in the United States for 2020 (Siegel et al., 2020). Currently, endocrine-based therapy has been practiced for advanced prostate cancer type though it does not show significant results in complete ablation of cancer. The metabolism of adrenal androgens in the prostate cancer cell is an important mechanism associated with the progression of castration-resistant prostate cancer (Banerjee et al., 2018). AKR1C3 is the important Aldo-keto reductase family member that plays a crucial role in the conversion of adrenal androgen dehydroepiandrosterone (DHEA) into high-affinity ligands for testosterone and dihydrotestosterone (Hamid et al., 2012). In human, AKR1C3 gene located in chromosome 10 which encode the enzyme, AKR1C3 is also known as 17β-hydroxysteroid dehydrogenase type 5 (17β-HSD5) involved in the biosynthesis of androgen (Penning, 2019). Hence, it acts like a potent drug target for prostate cancer treatment. The studies reported that AKR1C3 significantly induces abiraterone resistance via overexpression of intracrine androgen synthesis and improves the androgen signals and subsequently leads to the activation of the androgen receptor (AR) (Liu et al., 2017). The high expression leads to abiraterone resistance whereas the less expression of AKR1C3 leads to the high sensitivity of abiraterone in resistance cells. In prostate cancer, intracrine androgen levels are highly expressed hence, the treatment of these resistance cells results in a low level of intracrine androgen level and reduces the AR transcriptional activity subsequently increasing the efficiency of abiraterone therapy (Liu et al., 2015). Recently the studies have been recorded that tobacco smoking associated with prostate cancer and there is a shred of evidence that Aldo-keto reductase 1C3 enzymes involved in the metabolic activation of chemical carcinogens such as polycyclic aromatic hydrocarbons which are a rich source of tobacco (Karunasinghe et al., 2019). Besides AKR1C3 significantly catalyzes the extra-testicular androgen synthesis, therefore the process of tobacco metabolism is highly required AKR1C3. In the same studies, it was mentioned that four androgen pathways related genetic polymorphism (rs12529) is associated with a high risk of prostate cancer (Flück et al., 2011). This AKR1C3 rs12529 G allele, in particular, showed a negative association with serum PSA level when compared to healthy controls (Karunasinghe et al., 2019; Mansouri et al., 2020). The patients carrying variant allele G of rs12529 genetic polymorphisms can be detected in prostate cancer when interacting with environmental factors (Karunasinghe et al., 2017, 2019). The experimental studies reported the AKR1C3 rs12529 polymorphism can be an impenetrable variable for the prostate cancer patient stratification along with tobacco smoking status. The studies have been reported that single-nucleotide polymorphism in AKR1C3 is significantly linked to disease progression and aggressiveness in prostate cancer. In addition, several studies reported that the single nucleotide substitution in AKR1C1, AKR1C2 and AKR1C3 play a key role in intracrine androgen biosynthesis and metabolism. It highly induces the resistance of a number of anticancer drugs including oracin, cisplatin, and doxorubicin, and also it significantly elevated the protein levels which are associated with radio-resistance in lung cancer. Recently, phytocompounds have great attention due to no side effects and act as promising therapeutic candidates for the treatment of various types of cancers. For instance, the plant compound withaferin and its derivatives were used as a potentially effective alternative therapy against several types of cancers such as glioblastoma, lung, leukemia, and breast cancer via induction of apoptosis no studies reported that potent effective agents at the controlling of overexpression of AKR1C3 in prostate cancer (Khan et al., 2019). Hence in the current study, we performed in silico methods to find the deleterious SNPs and their corresponding genetic alterations in AKR1C3 and to investigate this genetic variation can alter the function of AKR1C3. Besides molecular docking, prime MM/GBSA and ADME were performed to analyze the molecular interaction of plant compounds with the AKR1C3 target receptor.
2 Materials and methods
The SNPs of AKR1C3 were retrieved from the Single-nucleotide Polymorphism (SNP) database and their related protein sequences in AKR1C3 were retrieved from Uniprot. The collected SNPs data were used for various In silico analysis.
2.1 Exploration of functional effects of coding nsSNPs by SIFT method
The alteration in the single amino acid at a certain position significantly affects the protein function, hence the prediction of single amino acid substitutions has great attention to insights the proteins and their implications in diseases. The genetic variants and mutations found in polymorphism sites that are responsible for complications are functionally and structurally important. Hence in this study, we used In silico tool SIFT program (Available at http://sift.jcvi.org/), which is used to predict and understand the mutation-associated polymorphism in AKR1C3 genes. SIFT is a multistep homology-based program that sorts tolerant amino acid substitution. Initially filter the closely related sequences that share similar functions and predict deleterious amino acid substitution at a particular position in a protein that may have a phenotypic effect (Ng and Henikoff, 2001). In the second step, it acquires the multiple alignments of the selected sequences and calculates the normalized probabilities of the amino acid substitutions at every position based on the multiple alignments and predicts the possibilities less than a cutoff value is deleterious (0.00 to 0.05); those have greater cutoff value are categorized as to be tolerated (above 0.05) (Hassan et al., 2019).
2.2 Prediction of functional changes on coding nsSNPs by Polyphen2
Identification of deleterious and damaged coding nsSNPs at the structural level is the important step to insights the functional changes in the protein. Polyphen2 is one of the commonly used programs for the probabilities of amino acid alterations and its impact on both structural and functional levels (Adzhubei et al., 2013). The amino acid sequence is utilized as input in a polyphen2 server with amino acid change and its position to characterize the polymorphism. It searches the three-dimensional structure and multiple alignments of similar amino acid sequences and mapping the substitution site which altogether is taken as a parameter by PolyPhen server and calculates the Position-Specific Independents Counts (PSIC) scores for each amino acids substitution and then computes the difference between the scores (Lopes et al., 2012). These scoring differences categorize the SNPs as probably damaging, potentially damaging, and damaging (Hepp et al., 2015). (Available at: http:// genetics.bwh.harvard.edu/pph).
2.3 Examining the stability change in mutation by I-mutant 2.0
To evaluate the structure–function relationships in SNPs were predicted by using I-mutant (Capriotti et al., 2005). It estimates the free energy changes by computing the Gibbs free energy for the wild type protein and subtracting it from a mutant form. The predicted values of all mutant types in AKR1C3 may change the protein stability of the AKR1C3 with related free energy. Four different outputs can be retrieved from the analysis (Younus et al., 2018). The positive DDG values indicate the high stability of the mutated proteins, whereas the negative score indicates lower stability. Available at http://gpcr2.biocomp.unibo.it/cgi/predictors/IMutant3.0/I-Mutant3.
2.4 PHD-SNP
PHD-SNP is a vector machine-based online tool widely used to predict the site-directed mutation in protein sequence and provide information about the deleterious amino acid substitutions and their impact on diseases (Capriotti and Fariselli, 2017). The output of this program calculates the reliability index score and predicts whether the single point amino acid change leads to diseases. Available at http://snps.biofold.org/phd-snp/phdsnp.html.
2.5 Analysis of amino acid evolutionary conservation
Consurf is used to analyze the evolutionary conservation of AKR1C3 amino acids. It determines the conserved amino acids with an empirical Bayesian method to identify the structural and functional residues in the conserved regions (Ashkenazy et al., 2016). Based on the conservation score and color indications it predicts the amino acids into a variable (range between 1 and 4), intermediate (range between 5 and 6), and conserved amino acids (range between 7 and 9) (Ashkenazy et al., 2010). Available at http://consurf.tau.ac.il/.
2.6 PROVEAN (Protein variation effect analyzer)
To predict the alterations in the biological functions of a protein owing to the single amino acid substitutions based on the sequence clustering and alignment scoring. A score less than −2.5 to be predicted as deleterious (Choi and Chan, 2015). Available at http://provean.jcvi.org/index.php.
2.7 Preparation of ligands and protein
Plant compounds reported with biological activity against prostate cancer were selected based on the literature. Around 30 compounds were selected, and the 2-dimensional structures of the compounds were initially retrieved from the PubChem database (Kim, 2016). The 3D conversion and minimization of the selected compounds were performed using the Ligprep module implemented in Maestro with the OPLS-2005 force field (Smak et al., 2021; Vijayalakshmi et al., 2013). Initially, 32 stereoisomers and possible ionizations were generated per ligand at pH of 7.0, using Confgen maximum of 1000 conformers per ligand were generated with default parameters (Shafreen et al., 2013). The 3D structure of AKR1C3 was retrieved from the PDB database (PDB ID: 6GXK) and subjected to protein preparation using the Protein Preparation Wizard script implemented in Maestro. Initially, all the missing residues were added and removed all the co-crystalized water molecules. The structure was optimized with contacts by changing the hydroxyl group orientations and flipping the side chain residues followed by energy refinement with the OPLS-AA force field (Selvaraj et al., 2014b).
2.8 ADME
ADME properties of the selected compounds were analyzed using the QikProp module (QikProp, version 3.8. Schrodinger LLC) of Schrodinger. Both physical and pharmaceutically relevant parameters are important descriptors to validate the drug-like properties of the compound (Muralidharan et al., 2015). The parameters such as Lipinski rule of five, number of hydrogen bond acceptor, donor, and molecular weight of the molecule, and logP values are some of the essential parameters which are required as basic features for drug-likeness of the molecules (Mansouri et al., 2020).
2.9 Molecular docking
Grid-Based Ligand Docking with Energetic (GLIDE) is one of the widely used techniques used to evaluate the molecular interaction of small molecules with their potential drug targets. In this study, the selected plant compounds are docked with both wild-type and mutated type AKR1C3 target proteins. Before the molecular docking, the binding site of the proteins was predicted by the SiteMap program and the receptor grid was generated with a grid box enclosing centered to the active site residues (Reddy et al., 2013). Then the molecular docking was performed with XP mode which performs via a conformational search for a ligand and also determines all the reasonable orientations for each ligand. The complete search of docked molecules provides the scoring function (G-score) to select the best conformation of the small molecules. The protein and ligands were adjusted and minimized up to 0.3 Å RMSD with OPLS-AA force field (Suryanarayanan et al., 2013). The hydrogen bonds, bond lengths, and hydrophobic interactions between AKR1C3 and plant compounds were determined by using PyMol and Maestro.
2.10 Binding energy calculation
The binding free energies of the ligand and receptor complexes of docked molecules were predicted using Prime MM/GBSA implemented in Schrodinger. In this process, two methods were combined such as the optimized potential for liquid simulation- all-atom (OPLS-AA) and molecular mechanics energy, an SGB solvation model for polar solvation (GSGB), and a nonpolar solvation term (GNP) composed of the nonpolar solvent accessible surface area and van der Waals interactions (Selvaraj et al., 2014a). Binding energy was calculated by the following formula: where, Ecomplex is the energy of the protein-inhibitor complex, Eprotein is the energy of protein and Eligand is the energy of ligand (Chinnasamy et al., 2020).
2.11 Molecular dynamics simulations
Structural analysis for the stability of ligands with the AKR1C3 receptor and the also the mutant apo state is carried out for MD simulation, trajectory analysis with the molecular dynamics simulation package – GROMACS 5.0 (Shafreen et al., 2013; Shree et al., 2020). For apo state, only protein is included in the system, and for ligand bound complex, the topology of ligand is included from PRODRG external server, followed by similar protocol of apo protein (Selvaraj et al., 2020a, 2020b). The orthorhombic box is filled with TIP3P solvent (water) molecules, measuring 2.5 nm from the protein and protein–ligand complex. For neutralizing the Na + and Cl- ions are added along with the solvent and steepest descent method is applied for energy minimization of the system (Choudhary et al., 2020; Sivakamavalli et al., 2016). Room temperature is set to 300 K v-rescale thermostat for the control of temperature coupling and for maintaining the pressure 1 bar, the Parrinello-Rahman with coupling constant is set to 2.0 ps (Sasidharan et al., 2020). For controlling the electrostatic and vdW interactions, the Partial Mesh Ewald (PME) is applied and bond length between the atoms are constrained by using the LINCS algorithm (Umesh et al., 2020). EM and NPT followed by NVT ensembles are simulated for 100 ps for equilibration and MD simulations are processed for 20 ns for understand the stability of apo protein and ligand bound complex (Muralidharan et al., 2015). Results of MD simulations are analyzed using the RMSD graph calculated based on reference initial position.
3 Results
3.1 nsSNP retrieval and function prediction by SIFT and PolyPhen method
A total of 18,594 SNPs in AKR1C3 gene were retrieved from dbSNP database, among them around 279 SNPs were predicted as non-synonymous, 136 were in non-coding region 3′ UTR and 5 were in 5′ UTR regions and rest of the SNPs were found in the intronic region. In the current study, we selected all the non-synonymous, 5′UTR, and 3′ UTR SNPs for computational analysis. Various computational tools such as SIFT, PolyPhen, I-mutant, PROVEAN, molecular docking, and ADME were used in this study to predict the deleterious nsSNPs. Among the 276 nsSNPs, only eight SNPs were found to be deleterious with the tolerance index score range from 0.00 to 0.009 (Table 1). From these eight SNPs, two nsSNPs had high tolerance scores 0.00, 2 had 0.001, and remaining SNPs showed 0.003, 0.004, 0.005 and 0.009. This low index of SIFT score values indicate the predicted nsSNPs are had low tolerance and may lead the functional changes in the amino acids sequence and also it may significantly affect the stability and functions of the proteins. The structural level changes in the protein sequences, the predicted nsSNPs were subjected to the PolyPhen server. A position-specific Independent Count score (PSIC score) was calculated and the difference of 1.5 and above were predicted to be damaging SNP. All the eight nsSNPs show a PSIC score ranging from 0.666 to 1.000 was shown in Table 1.
SNP ID
SIFT Score
Amino acid change
Tolerance
PSIC score
DDG
rs11551177
0.009
E54G
DELETERIOUS
0.946
−0.34
rs62621365
0.005
R258C
DELETERIOUS
0.000
−0.48
rs139011578
0.001
A52G
DELETERIOUS
1.000
−1.23
rs145644085
0.000
V111D
DELETERIOUS
1.000
−1.42
rs199934766
0.000
R199W
DELETERIOUS
0.666
−1.25
rs370390057
0.004
Y296S
DELETERIOUS
0.787
−1.88
rs370769193
0.003
K247N
DELETERIOUS
1.000
−0.15
rs377392226
0.001
A52P
DELETERIOUS
0.918
−0.77
3.2 Stability analysis of I-mutant
Out of eight nsSNPs, I-mutant server predicts the two non-synonymous SNPs such as rs199934766 and rs11551177 showed a DDG value of >−1.0. The remaining deleterious SNPs such as rs62621365, rs139011578, rs145644085, rs370390057, rs370769193, and rs377392226 have less DDG score (<−1.0). The support vector machine (I-Mutant) effectively predicts the change of protein stability free energy (ΔΔG or DDG) on a specific nsSNP. The high negative score DDG score indicates highly deleterious, except for two nsSNPs such as rs199934766 and rs11551177, the remaining four nsSNPS were predicted as less stable than the given point mutation.
3.3 PHD-SNP and project HOPE
PHD-SNP is the widely used tool used for the prediction of diseases associated with nsSNP. All the eight deleterious SNPs were subjected to PHD-SNP analysis and it was predicted that all the eight SNPs were found to be diseases related polymorphism (Table 2). The amino acid substation of the deleterious SNPs was predicted by HPOE software. Based on the results of other SNP prediction tools such as SIFT, Poly Phen, I mutant, nsSNP rs62621365 was depicted as most deleterious and damaging hence this single nsSNP alone was selected and subjected to HOPE software. The results obtained from the HOPE server depicted that the mutant residue is smaller than wild-type residues hence the mutated amino acid may contact with structural domains and the mutant residues was not in the other residues types observed at this position this may due to some rare case mutation which occurs in the protein (Fig. 1 & Fig. 2.).
SNP ID
Amino acid change
PROVEAN score
Prediction (Cutoff = -2.5)
PhD-SNP (PhD-Predictor of human Deleterious)
rs11551177
E54G
−5.777
Deleterious
Neutral, Disease-related Polymorphism
rs62621365
R258C
−7.238
Deleterious
Neutral (6) Disease-related Polymorphism
rs139011578
A52G
−3.596
Deleterious
Neutral (1) Disease-related Polymorphism
rs145644085
V111D
−6.259
Deleterious
Neutral (1) Disease-related Polymorphism
rs199934766
R199W
−3.043
Deleterious
Neutral 7 Disease-related Polymorphism
rs370390057
Y296S
−3.840
Deleterious
Neutral 4 Disease-related Polymorphism
rs370769193
K247N
−3.879
Deleterious
Neutral 5 Disease-related Polymorphism
Rs377392226
A52P
−4.533
Deleterious
Disease 9 Disease-related Polymorphism
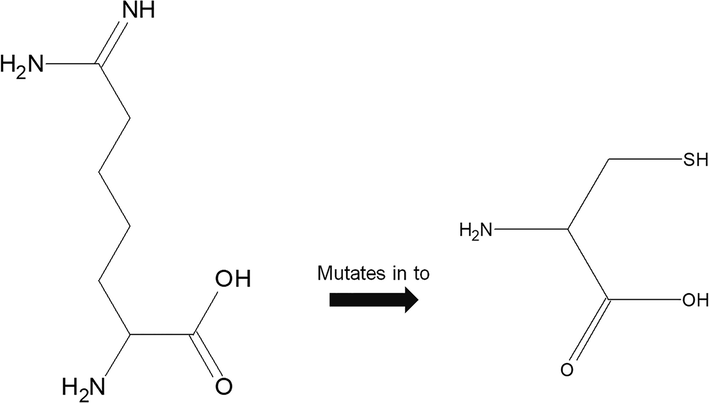
Shows the schematic structure of the original (left) and mutant (right) amino acid predicted by HOPE software.
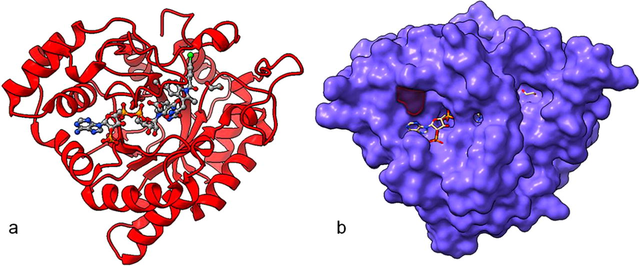
Shows the AKR1C3 protein with a mutated amino acid. Proteins are represented in red color in ribbon model (a) and purple surface enabled with highlight of mutant in red color (b).
3.4 Analysis of amino acid evolutionary conservation and PROVEAN
The evolutionary changes in the amino acid sequence and evolutionary conservation profile of the AKR1C3 gene were examined using the ConSurf server which uses the Bayesian method to detect the evolutionary profile of the gene and also determine the evolutionarily conserved amino acids in proteins. Fig. 3 shows the predicted results of ConSurf of AKR1C3 based on the conserved residues; there are 9 color variations and conservation scale of each residue which indicates the evolutionary relationship. In Fig. 3, it was noticed that conservation scale e, b, f, and s; where e; represent the exposed residue, f represent the highly conserved residues, b; indicated the buried residues and s; represent the highly conserved structural residues, and f; indicates the highly conserved functional residues. The color variation ranges from light to dark purple indicate the scale of conserved region, from the Fig. 3 it was noticed that the important amino acids that were involved in binding mechanism such as Tyr216, Leu268, Leu219, Asn167, Tyr24, Asp50, Tyr55, Phe311 were in dark purple color region which indicates the most of the binding pocket amino acids were present in conserved region. Further, the PROVEAN results also predict the impact of deleterious SNPs may cause significant biological function in protein. All the eight nsSNPs of AKR1C3 were predicted as deleterious and the rs62621365 shows highly deleterious with PROVEAN score −7.238 (Table 2).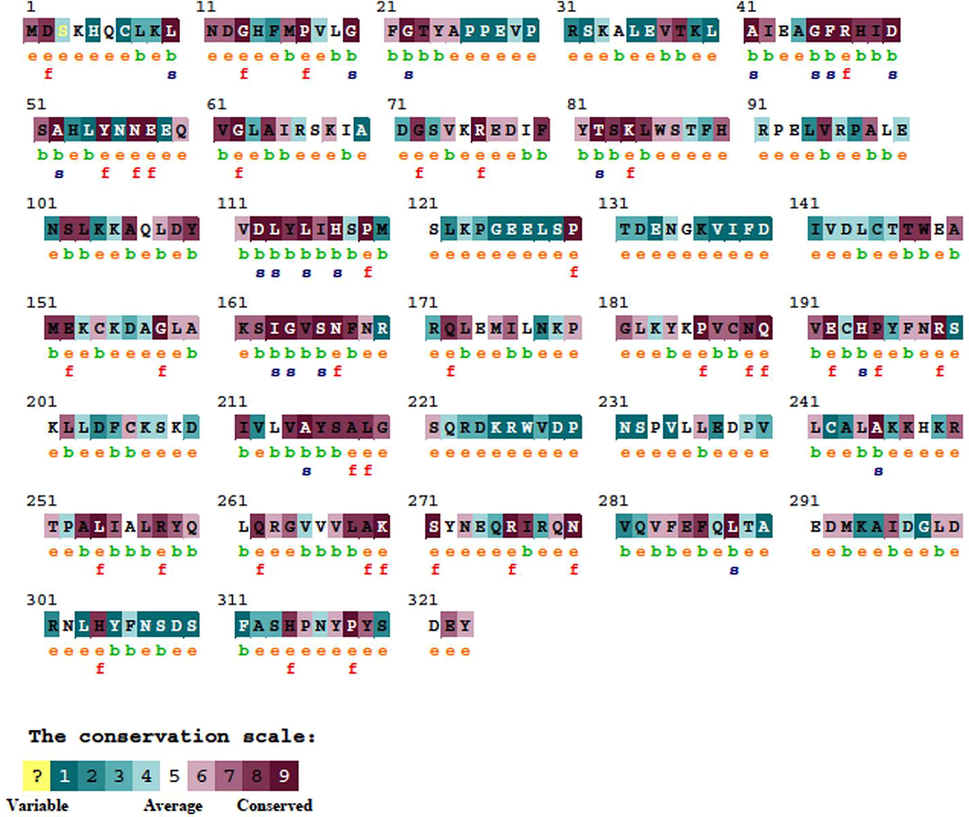
Evolutionary conserved amino acid profile of AKR1C3 retrieved from ConSurf. The color-coding bar shows the conservation score.
3.5 ADME
In the drug designing process, the prediction of small molecule scaffolds with good binding affinity and potent ADME properties is the main criteria. In this study, ADME properties of all the selected plant compounds were predicted using the QikProp module. Basic significance of both physical and pharmaceutically relevant properties was analyzed. The significant parameters such as QPlogBB, logP, (Octanol/Water), QPlogS, APPCaco, and permeability via Madin-Darby Canine Kidney (MDCK) cells in mm/sec, Lipinski's rule of five, molecular weight, and human oral absorption were examined for this study and values are shown in table 3. The partition coefficient (QplogPo/w) and the QPlogS are important criteria and essential for evaluating the absorption and distribution of drug molecules in the body. The values calculated for QPlogPo/w and QPlogS (water solubility) range from −0.161 to 4.564 and −2.883 to −8.067 respectively. The brain barrier coefficients of all the plant compounds range from 0.243 to 107.54, the value of the permeability via MDCK cells significantly mimics the blood–brain barrier. a Predicted apparent MDCK cell permeability in nm/s (mimic for brain/blood barrier) (<25 is poor, >500 great. b Predicted apparent Caco2 cell permeability in nm/s (<25 is poor, >500 great). c Predicted brain/blood partition co-efficient; logBB (acceptable range 3.0 to 1.2). d Predicted aqueous solubility; S in mol/l (acceptable range 6.5 to 0.5). e QPlogPo/w: Partition coefficient; recommended range 2.0 to 6.5. f Predicted percentage of human oral absorption (<25% is poor). g Number of property or descriptor values that fall outside the 95% range of similar values for known drugs (range: 1–5).
Compound Name
QPPMD CKa
QPlog HERG
QPPCacob
QPlog BBc
QPlog Sd
QPlog Po/we
M.W
Rule of fiveg
% of oral absorbance
Ginkgetin
7.987
−7.379
21.98
−3092
−8.067
4.564
566.50
1
64
Withaferin A
107.54
−4.493
243.68
−1.340
−4.961
2.97–3
470.60
0
87
Resveratrol
0.243
−5.89
0.867
−4.509
−3.699
−0.259
458.40
2
0.000
Genistin
8.300
−5.81
22.782
−2.833
−2.935
−0.161
432.40
1
37
3.6 Molecular docking interaction analysis
The molecular interaction of protein–ligand complex plays an essential role in the structure-based drug discovery process. The molecular docking of plant-based compounds with AKR1C3. The binding energies and interaction residues of the best four compounds with both wild type and mutant form of AKR1C3 were tabulated in table 4. A total of 30 plant compounds that show potent anticancer activity were selected for this study among them Ginkgetin, Withaferin A, resveratrol, and Genistin show the best scoring and binding energy with AKR1C3 receptor. These results suggested that Ginkgetin shows the greatest binding energy (-13.326 k/cal), and forms hydrogen bond interaction and π-π interaction with Tyr216 and Phe360, respectively. Followed by Ginkgetin, Withaferin A (-10.620 k/Cal), Resveratrol (-10.086 k/Cal) shows the more or less similar binding score and glide energy. Molecular interaction analysis (Fig. 4) depicted that the best four bioactive compounds with wild type AKR1C3 showed good interaction with its active site residues. The binding mode of Ginkgetin depicted that it makes interaction with binding site residues via π-π interaction with Tyr216, Phe306. Another compound Withaferin A forms hydrogen bond interaction with Lue268 via –O- and –OH– atoms (O…H, CO.. Leu268) with a distance of 2.09 Å and Leu219 via –N– and –OH– (O…H, NH… Leu219) with a distance of 2.42 Å and Asn167 via –CH– and –NH– (C…H, NH… Asn167) with distance of 2.03 Å, Resveratrol makes two hydrogen forms with Asp50 via –O– and –OH– (C…O, OH…Asp50) with distance of 1.67 and Tyr24 via –O– and –NH– (O…H, NH…Tyr24) with distance of 2.20 Å, and π-π interaction with Trp227, Tyr24. Genistin forms hydrogen bond interaction with Asp50 via –C– and OH (C…O, HO…Ap50) with distance of 2.50 Å, and two hydrogen bonds with Asn167 via –N–, –OH– (N…H, OC, Asn167) with distance of 2.69 Å, and 2.38 Å, and with Tyr55 via –H– and –OH– (H…O, HO… Tyr55) with a distance of 2.12 Å, in addition it also forms π-π interaction with Phe311, Phe306. Fig. 5 represents the molecular interaction analysis of mutant form of AKR1C3 with four plant compounds. From the Fig. 5 it was observed that all the four plant compounds showed more or less similar docking score and also similar interactions compared to wild type, Ginkgetin forms four hydrogen bond interaction with Asn167 via –N– and –CO– (N..H, OC…As167) with distance of 1.92 Å, and Asp50 via –O– and –OH– (C…O, OH…Asp50) with distance of 1.98 Å, Tyr24 via –N– and –OC– (N…H, OC…Asp24) with distance of 2.44, and forms hydrogen bond with Ser217 via –O– and –NH– (C…O, NH…Ser217) with distance of 1.97 Å, Withaferin A form three hydrogen bonds with Gln192 via –O– and –OH– (C…O, OH…Gln192) with distance of 2.29 Å, and Ser219 via –O– and –OH– (C…O, OH…Ser219) with distance of 2.18 Å and forms three π–π interaction with Tyr216. Resveratrol form single hydrogen bond interaction Ser217 via –O– and OH (O…H, OC…Ser217) with distance of 1.92 Å, and Genistin forms hydrogen bond interaction with Ser217 via –O– and –OH– (O…H, OC…ser217) with distance of 2.42 Å, Leu268 via –O– and OH… (C…H, OH…Leu268) with distance of 2.40 Å and Lys270 via –N– and –OH– (N…H, OC…Lys270) with distance of 2.15 Å and also forms π-π interaction with Trp227.
S. No
Compound name
Glide score (Kcal/mol)
Glide energy (Kcal/mol)
MMGBSA ΔG-bind energy (Kcal/mol)
Interaction residues
Wild type
Mutant
Wild type
Mutant
Wild type
Mutant
1
Ginkgetin
−13.326
−12.239
−96.350
−96.350
−79.26
Tyr216, Phe306 (π-π)
Asn167, Phe306(π-π), Asp50, Tyr24
2
Withaferin A
−10.620
−11.272
−97.896
84.410
−92.30
Leu268, Leu219, Asn167
Tyr216 (3 π-π), Glu192
3
Resveratrol
−10.086
−10.342
−82.945
−85.311
−55.38
Trp227, Tyr24 (π-π), Asn167, Tyr55, Asp50
Gln222, Ser217
4
Genistin
−9.239
−8.305
−72.592
−67.293
−74.16
Phe311, Phe306, (π-π), Asp50, Asn167, Tyr55
Trp227 (π-π), Ser217, Leu268
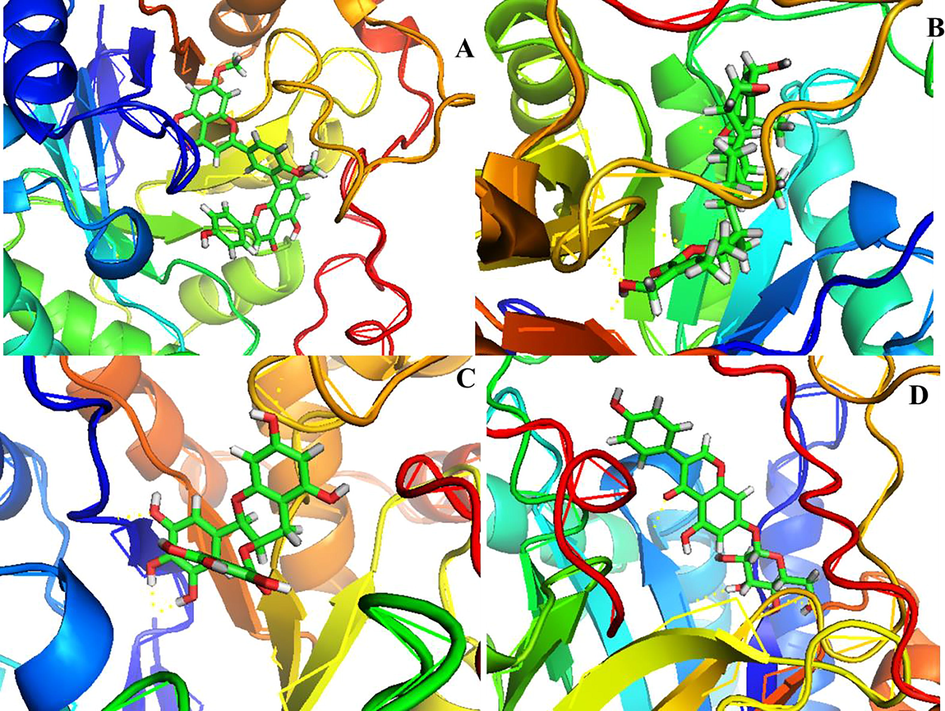
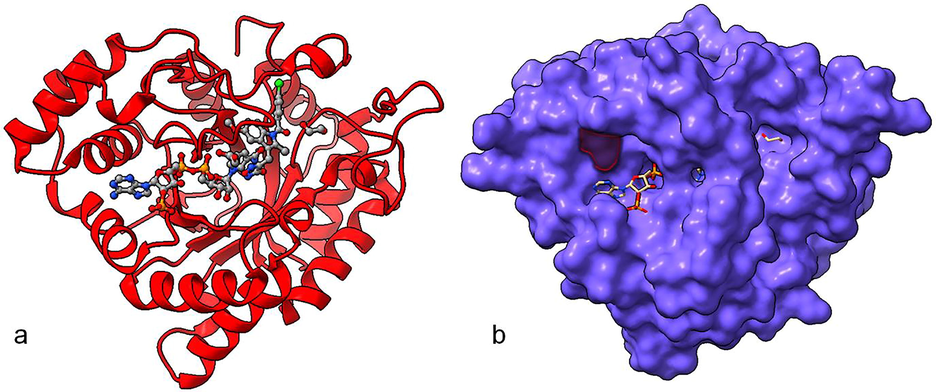
Represent the molecular interaction of the best-docked plant compounds Ginkgetin (A), Withaferin A (B), Resveratrol (C) and Genistin (D).
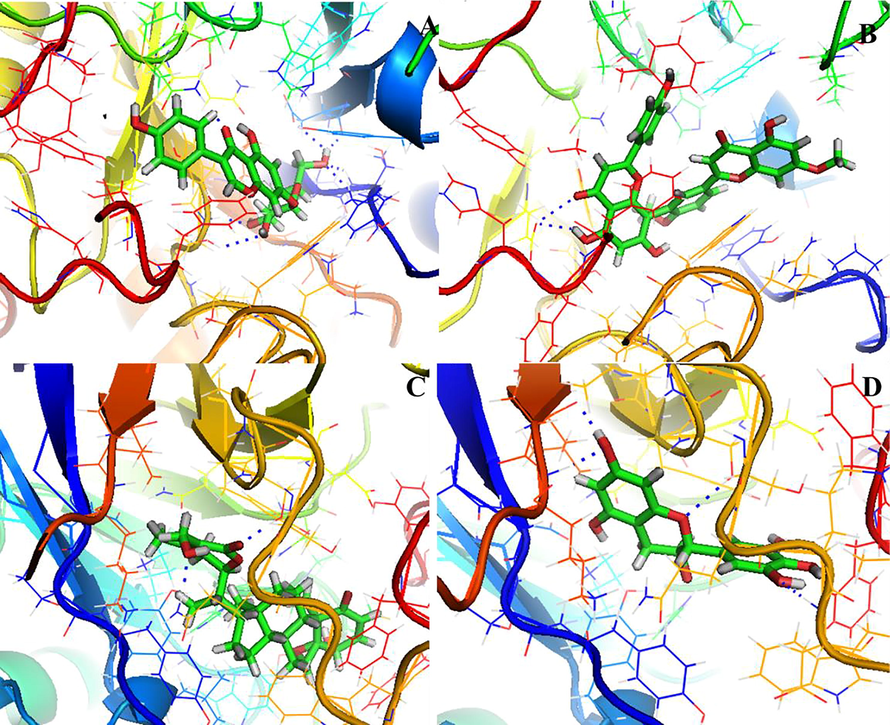
Represent the molecular interaction of the best-docked plant compounds Ginkgetin (A), Withaferin A (B), Resveratrol (C) and Genistin (D) with mutant form of AKR1C3 (A258C).
3.7 Binding energy calculation
The calculation of binding energies provides the more reliable binding affinity of the complex than molecular docking and also enhances the scoring functions of the docked complex for the ordering of the docked poses. The prime MM/GBSA calculation of the results in the best complexes was given in Table 3. The low and negative binding energy of all the complexes strongly indicated the all the plant compounds are in the most favorable conformation. The ligand-binding complex of all the best compounds ranged from −55.38 to −92.30 kcal/mol (Table 4). This low energy of the complex may be contributed by the other energies including van der Waals, h-bond, and electrostatic interactions.
3.8 Molecular Dynamics Simulations
For getting the stability of the mutant form of protein and protein ligand interactions, the MD simulation is carried out for the timescale of 20 ns for getting the RMSD values difference from its original position (Selvaraj et al., 2018). RMSD values from 20 ns of MD simulations are plotted against the initial position and provided in the Fig. 6, showing the RMS deviations for apo protein and also for the four-ligand bound complex. The simulation data shows that, for the whole 20 ns of MD simulations, the protein is stable inside the system by showing the RMSD values between ∼0.3 to 0.4 nm and for the ligand bound complexes, the RMSD lies between the ∼0.3 to 0.6 nm. This is clearly shown in the Fig. 6 represent the stability in the MD simulations for both apo and ligand bound complex. None of ligand bound complex loss its stability in the 20 ns of timescale represents the compounds are well bound inside the active site and stable inside the protein, by means of its bonding interactions.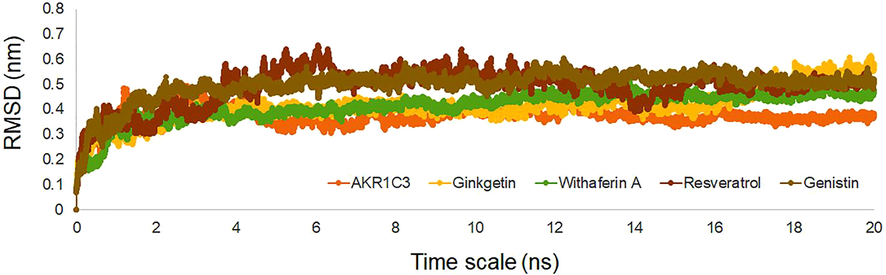
Molecular dynamics simulations of apo form of AKR1C3 along with Ginkgetin, Withaferin A, Resveratrol and Genistin bound complex showing the RMSD values for the timescale of 20 ns.
4 Discussion
From the SNP results available in the table 1, it shows that eight nsSNPs were predicted to be deleterious from the SIFT program and the PolyPhen analysis showed that all were found to be probably damaging. Hence, we could infer the predicted results from both PolyPhen and SIFT which specifies the structural details. Stability analysis prediction shows that the nsSNPs rs62621365 and rs145644085 were seen to be less stable, deleterious, and found damaging. PHD-SNP program-based results shows that, both wild-type and mutant type residues form hydrogen bonds with Glu262 and Val283. Also, the difference in hydrophobicity is noted in the site and it may affect the hydrogen bond formation in the mutant conditions. Depending on the homology and similarity of the proteins it calculates the evolutionary profile and predicts the substitution of the deleterious effect. From the results obtained from the Consurf, it was observed that most of the amino acids are present in conserved regions. Consurf result also shows that the highly deleterious SNP and the AKR1C3 variant A258C have the conserved scale 8 and the residue was located in the highly conserved functional residue domain hence the Consurf results also indicates that the rs62621365 mutation may make important functional residue changes in protein. In terms of ADME physiochemical property analysis, all the best four compounds follow the rule of five and human oral absorption and other parameters within the desirable range. The present results indicate that all the bioactive compounds follow all the ADME properties within the acceptable range. Based on the docking score and energy scores, the best four compounds are subject to molecular docking with mutant form of AKR1C3 (A258C). Molecular docking results shows that the taken plant compounds may act as a potent inhibitor for both wild and mutant (A258C) with more or less similar binding mode of interactions. For conformations, MM/GBSA based binding energy calculations are executed, and results revealed that the plant compound shows a potent binding affinity with the AKR1C3 receptor. For confirming its efficiency in the dynamic state of the mutant protein and complex system with plant compounds, the MD simulations for 20 ns provides the support for the interactions and binding. MD simulations shows both apo and ligand bound complex are stable inside the system. There is lack of high fluctuations seen in both apo and ligand complex shows, that ligands are well bound inside the active site and also shows prominent binding feature helps the ligand to stay inside the pocket, even in the dynamic state. The overall study concluded that the in-silico prediction and molecular interaction studies suggested that rs62621365 was considered as the most deleterious nsSNP and the plant compounds such as Ginkgetin and Withaferin A may act as potent inhibitors that can effectively suppress the level of AKR1C3 expression in prostate cancer.
5 Conclusion
AKR1C3 is reported as a potential biomarker to identify prostate cancer and the high expression of AKR1C3 levels may amenable to a novel form of hormonal treatment and it can convert the androgen into active growth-stimulating androgen which interfering with the AKR1C3 protein activity. In this study, we selected several nsSNP of AKR1C3 and most of the nsSNPs are non-coding regions. The structural analysis of selected high-risk nsSNPs predicted that the amino acid substitutions in AKR1C3 had a deleterious impact on the stability and function of the protein. The amino acid residues such as R258C (rs62621365) and V111D (rs145644085) undergo mutation. Molecular docking and ADME and MM/GBSA calculation revealed that the selected plant-derived compounds show good binding energy and molecular interaction with the AKR1C3 receptor. The overall results depicted that nsSNPs may be considered for risk assessment against prostate cancer.
Acknowledgement
The author would like to thank the Deanship of Scientific Research at Majmaah University, Al Majmaah, 11952, Saudi Arabia for supporting this work under Project Number No. R-2021-148.
Declaration of Competing Interest
The author declares that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. Chapter. 2013;7(Unit7):20.
- [Google Scholar]
- ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res.. 2016;44(W1):W344-W350.
- [Google Scholar]
- ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res.. 2010;38(Web Server):W529-W533.
- [Google Scholar]
- Androgen action in prostate function and disease. Am. J. Clin. Exp. Urol.. 2018;6:62-77.
- [Google Scholar]
- PhD-SNPg: a webserver and lightweight tool for scoring single nucleotide variants. Nucleic Acids Res.. 2017;45(W1):W247-W252.
- [Google Scholar]
- I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res.. 2005;33(Web Server):W306-W310.
- [Google Scholar]
- Regulation of aldo-keto reductases in human diseases. Front. Pharmacol.. 2012;3:35.
- [Google Scholar]
- Combining in silico and in vitro approaches to identification of potent inhibitor against phospholipase A2 (PLA2) Int. J. Biol. Macromol.. 2020;144:53-66.
- [Google Scholar]
- PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31(16):2745-2747.
- [Google Scholar]
- Understanding the biological role of PqqB in Pseudomonas stutzeri using molecular dynamics simulation approach. J. Biomol. Struct. Dyn. 2020:1-13.
- [Google Scholar]
- Androgen receptor structure, function and biology: from bench to bedside. Clin Biochem Rev. 2016;37:3-15.
- [Google Scholar]
- Why boys will be boys: two pathways of fetal testicular androgen biosynthesis are needed for male sexual differentiation. Am. J. Hum. Genet.. 2011;89(2):201-218.
- [Google Scholar]
- Aldo-keto reductase family 1 member C3 (AKR1C3) is a biomarker and therapeutic target for castration-resistant prostate cancer. Mol. Med.. 2012;18(11):1449-1455.
- [Google Scholar]
- Evaluation of computational techniques for predicting non-synonymous single nucleotide variants pathogenicity. Genomics. 2019;111(4):869-882.
- [Google Scholar]
- Prediction of the damage-associated non-synonymous single nucleotide polymorphisms in the human MC1R gene. PLoS ONE. 2015;10(3):e0121812.
- [Google Scholar]
- Influence of aldo-keto reductase 1C3 in prostate cancer – a mini review. Curr. Cancer Drug Targets. 2017;17:603-616.
- [Google Scholar]
- Interaction between leukocyte aldo-keto reductase 1C3 activity, genotypes, biological, lifestyle and clinical features in a prostate cancer cohort from New Zealand. PLoS ONE. 2019;14(5):e0217373.
- [Google Scholar]
- Anticancer plants: a review of the active phytochemicals, applications in animal models, and regulatory aspects. Biomolecules. 2019;10(1):10.
- [Google Scholar]
- Getting the most out of PubChem for virtual screening. Expert Opin. Drug Discov.. 2016;11(9):843-855.
- [Google Scholar]
- Inhibition of AKR1C3 activation overcomes resistance to abiraterone in advanced prostate cancer. Mol. Cancer Ther.. 2017;16(1):35-44.
- [Google Scholar]
- Intracrine androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer Res.. 2015;75(7):1413-1422.
- [Google Scholar]
- Maternal and offspring genetic variants of AKR1C3 and the risk of childhood leukemia. Carcinogenesis. 2008;29(5):984-990.
- [Google Scholar]
- A combined functional annotation score for non-synonymous variants. Hum. Hered.. 2012;73(1):47-51.
- [Google Scholar]
- CoMPARA: collaborative modeling project for androgen receptor activity. Environ. Health Perspect.. 2020;128(2):027002.
- [CrossRef] [Google Scholar]
- Structure-based virtual screening and biological evaluation of a calpain inhibitor for prevention of selenite-induced cataractogenesis in an in vitro system. J. Chem. Inf. Model.. 2015;55(8):1686-1697.
- [Google Scholar]
- AKR1C3 (type 5 17beta-hydroxysteroid dehydrogenase/prostaglandin F synthase): Roles in malignancy and endocrine disorders. Mol. Cell. Endocrinol.. 2019;489:82-91.
- [Google Scholar]
- Identification of potential HIV-1 integrase strand transfer inhibitors: in silico virtual screening and QM/MM docking studies. SAR QSAR Environ. Res.. 2013;24(7):581-595.
- [Google Scholar]
- Bacterial protein azurin and derived peptides as potential anti-SARS-CoV-2 agents: insights from molecular docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2020:1-16.
- [Google Scholar]
- Selvaraj, C., Dinesh, D. C., Panwar, U., Boura, E., Singh, S. K., 2020a. High-Throughput Screening and Quantum Mechanics for Identifying Potent Inhibitors against Mac1 Domain of SARS-CoV-2 Nsp3. IEEE/ACM Trans Comput Biol Bioinform PP.
- Microsecond MD simulation and multiple-conformation virtual screening to identify potential anti-COVID-19 inhibitors against SARS-CoV-2 main protease. Front. Chem.. 2020;8 595273
- [Google Scholar]
- Molecular dynamics simulations and applications in computational toxicology and nanotoxicology. Food Chem. Toxicol.. 2018;112:495-506.
- [Google Scholar]
- Virtual screening of LPXTG competitive SrtA inhibitors targeting signal transduction mechanism in Bacillus anthracis: a combined experimental and theoretical study. J. Recept. Signal Transduct. Res.. 2014;34(3):221-232.
- [Google Scholar]
- Structural elucidation of SrtA enzyme in Enterococcus faecalis: an emphasis on screening of potential inhibitors against the biofilm formation. Mol. BioSyst.. 2014;10(7):1775-1789.
- [Google Scholar]
- Exploration of fluoroquinolone resistance in Streptococcus pyogenes: comparative structure analysis of wild-type and mutant DNA gyrase. J. Mol. Recognit.. 2013;26(6):276-285.
- [Google Scholar]
- Targeting COVID-19 (SARS-CoV-2) main protease through active phytochemicals of ayurvedic medicinal plants - Withania somnifera (Ashwagandha), Tinospora cordifolia (Giloy) and Ocimum sanctum (Tulsi) - a molecular docking study. J. Biomol. Struct. Dyn. 2020:1-14.
- [Google Scholar]
- Modeling of macromolecular proteins in prophenoloxidase cascade through experimental and computational approaches. Biotechnol. Appl. Biochem.. 2016;63(6):779-788.
- [Google Scholar]
- Pan-selectin inhibitors as potential therapeutics for COVID-19 treatment: in silico screening study. Glycobiology 2021
- [Google Scholar]
- A three-dimensional chemical phase pharmacophore mapping, QSAR modelling and electronic feature analysis of benzofuran salicylic acid derivatives as LYP inhibitors. SAR QSAR Environ. Res.. 2013;24(12):1025-1040.
- [Google Scholar]
- Umesh, Kundu, D., Selvaraj, C., Singh, S. K., Dubey, V. K., 2020. Identification of new anti-nCoV drug chemical compounds from Indian spices exploiting SARS-CoV-2 main protease as target. J. Biomol. Struct. Dyn., 1–9.
- Exploration of the binding of DNA binding ligands to Staphylococcal DNA through QM/MM docking and molecular dynamics simulation. J. Biomol. Struct. Dyn.. 2013;31(6):561-571.
- [Google Scholar]
- Structure-function mutational analysis and prediction of the potential impact of high risk non-synonymous single-nucleotide polymorphism on poliovirus 2A protease stability using comprehensive informatics approaches. Genes (Basel). 2018;9(5):228.
- [CrossRef] [Google Scholar]




