

Translate this page into:
Identification of potential riboflavin synthase inhibitors by virtual screening and molecular dynamics simulation studies
⁎Corresponding author at: Department of Cell and Molecular Biology, Faculty of Biotechnology and Biomolecular Sciences, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia. adamleow@upm.edu.my (Thean Chor Leow)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
Riboflavin synthase is an important enzyme catalyzing the last step of riboflavin biosynthesis in microorganisms. Due to the absolute dependency of the microbes to this biosynthetic pathway coupled with its nonexistence in humans, riboflavin synthase (RiS) is considered as a prospective drug target. The riboflavin synthase for this study was derived from Leptospira kmetyi, a pathogenic bacterium locally isolated in Malaysia. Leptospirosis, an infectious disease caused by pathogenic Leptospira, is a serious growing public health issue. Treatment of leptospirosis with antibiotics over time resulted in the evolution of antibiotic resistance strains, thus requiring the development of newer but safe antimicrobial agents. In this study, a computational approach involving virtual screening followed by molecular dynamics (MD) simulation was implemented in order to identify possible inhibitors against riboflavin synthase. The model of Leptospira kmetyi riboflavin synthase predicted from E. coli riboflavin synthase (1I8D) was used as the drug target to screen for potential compounds from the ZINC database through virtual screening. The potential compound with the highest Glide score (−10.987 Kcal/mol) was identified to be ZINC21883831. Chemically it is 2-[(2-chloro-4-fluoro-phenyl)methylsulfanyl]-7-phenyl-3,5-dihydropyrrolo[2,3-e]pyrimidin-4-one. The top three docked complexes from the virtual screening and apo-structures were subjected to molecular dynamics simulation to predict the stabilities of the Leptospira kmetyi riboflavin synthase-ligand complexes. Stability parameters including RMSD, RMSF, SASA and Rg of the complexes were evaluated from 60 ns of the MD simulation trajectories. Insights from this study provide promising starting points for the rational designs of new effective and safe anti-leptospirosis drugs.
Keywords
Riboflavin synthase
Virtual screening
Molecular dynamics simulation
Leptospirosis

1 Introduction
Leptospirosis is an infectious disease threatening human and animal health and is regarded as an ubiquitous zoonosis (Victoriano et al., 2009). However, leptospirosis is an underreported disease, and worldwide leptospirosis incidence statistics are not reliable. According to the Leptospirosis Burden Epidemiology Group (WHO) estimation, there were 48,600 deaths out of 873,000 cases of leptospirosis reported annually. According to Costa et al. (2015), high morbidity and mortality rates were recorded in resource poor tropical countries. Antibiotics such as IV C-Penicillin (for severe adult patients), doxycycline, tetracycline, ampicillin or amoxicillin (for less severe cases) are the only medications to combat leptospirosis (Hakim, 2011). As antibacterial drugs have been used around the world for several decades, the emergence of drug-resistant bacteria poses severe health treats to human. Drug-resistant bacteria make the treatment of infectious diseases in hospitals more difficult and increase the rate of mortality in patients. Therefore, there is a need to take preventive actions for overcoming this situation before it becomes a major public health issue. The identification of novel drug targets for infectious bacteria such as Leptospira is crucial to combat antibiotic resistance in Leptospira species. Many metabolic pathways such as the citrate cycle, fatty acid metabolism and the electron transport chain depend on the coenzymes flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD). The precursor riboflavin (vitamin B2) for these coenzymes is synthesized by seven distinct enzymatic reactions from guanosine triphosphate (GTP). The endogenous riboflavin biosynthetic pathway supplies bacteria with riboflavin for cell metabolisms (Angulo, 2017; Bonomi et al., 2010; Zylberman et al., 2006). Riboflavin synthase (RiS) is the key enzyme in the riboflavin biosynthesis pathway that catalyzes the dismutation of two molecules of 6,7-dimethyl-8-ribityllumazine (DMRL) at the last step of the pathway to produce riboflavin and 5-amino-6-ribitylamino-2,4(1H,3H)-pyrimidinedione (Long et al., 2012). With the absence of riboflavin synthase in humans and the dependency of pathogenic bacteria towards the riboflavin biosynthesis pathway, RiS is therefore a prospective target for the designing of antimicrobial drugs (Serer et al., 2014; Dorrazehi et al., 2016). The riboflavin synthase gene was found in the genome of the pathogenic Leptospira kmetyi previously isolated from soil in Johor, Malaysia (Slack, et al., 2009). Leptospirosis is easily transmitted by contact with the urine of infected rodents at recreation and flooded areas (Garba et al., 2017). In addition, L. kmetyi was detected in the blood samples of patients suffering from acute leptospirosis (Bourhy, et al., 2013). These discoveries make it a potential drug target. The Structure-based Drug Design (SBDD) technique expedites the lead discovery process by pharmaceutical companies and scientists at reasonable costs. It is a precise and effective way for discovering potential leads which act on drug targets at the molecular level. The SBDD technique acts as a virtual shortcut and can reduce the cost of research and development (Leelananda and Lindert, 2016). Amprenavir is a potential HIV protease inhibitor which was discovered by the in silico protein modeling and molecular dynamics (MD) simulation approaches (Clark, 2006). Other inhibitors such as Raltitrexed for thymidylate synthase; the antibiotic Norfloxacin for urinary tract infection and Isoniazid for tuberculosis were discovered via structure based virtual screening methods (Rutenber and Stroud, 1996). The virtual screening method is used to identify potential leads from compound libraries targeting a specific enzyme or receptor (Gimeno et al., 2019). MD simulation is very useful in investigating the pathogenic mechanism of a disease, and the stability of a protein–ligand interaction both of which are difficult to solve experimentally. MD simulations provide dynamical structural information on biomacromolecules for unveiling the structure–function relationship of protein–ligand interaction towards drug discovery (Liu et al., 2017). An in silico approach study by Ibrahim et al. (2020), revealed the active compound for the design of a new anti-diabetic drug against α-glycosidase. In this study, the combined strategies of molecular modelling, virtual screening and MD simulations were used to reveal some potentially promising drugs to be used against leptospirosis.
2 Materials and methods
2.1 Protein preparation
As the crystal structure of riboflavin synthase from Leptospira kmetyi (hereafter referred as LkRS) is not yet reported, homology modelling was performed to predict the LkRS structure. The primary sequence of locally isolated LkRS was obtained from the NCBI (National Center for Biotechnology Information) under the Accession no: WP 010575348.1. The PSI-BLAST analysis of LkRS against PDB revealed a crystal structure of riboflavin synthase from E. coli (1I8D) as the closest homolog, sharing 39% identity. Prediction of the LkRS structure was conducted using the Yet Another Scientific Artificial Reality Application (YASARA) software (Krieger et al., 2002) with the crystal structure of riboflavin synthase from E. coli (1I8D) as a template. After the side-chains had been built, optimized and fine-tuned, all the newly modelled parts were subjected to a combined steepest descent and simulated annealing minimization procedure. Simple minimization was carried out using AMBER03 force field and the model was refined in explicit solvent molecules. The model with the highest quality score was chosen as the final 3D structure model and the quality was evaluated by the Procheck module of the PDBsum server (Laskowski et al., 2005) and ProSA analysis (Wiederstein and Sippl, 2007). The validated model was optimized and refined using the Maestro software’s protein preparation wizard of GLIDE (Glide, Schrödinger, LLC, New York, NY, USA). Optimization of the protein such as bond orders, ionization states, steric clashes and side chain orientations for PDB files were corrected and the energy minimized for all atoms using force field Optimized potential for liquid simulations 2005 (OPLS) prior to virtual screening studies.
2.2 Ligand preparation
One million commercially available drug-like compounds were shortlisted and retrieved in .sdf format from the ZINC database (http://zinc.docking.org) by applying Lipinski’s rule (Lipinski, 2004) of five. All of these compounds were subjected to ligand preparation using the Ligprep module of the Schrodinger Suit. During the process, tautomeric states and ionization states were generated using the Epic module. Furthermore, appropriate charges were assigned, ligands were neutralized and energy minimization performed. Finally, the low energy ring conformations of all the ligands were generated and then these prepared ligands were further used for the virtual screening.
2.3 Grid preparation
The Glide's receptor grid generation wizard was used after the protein preparation to produce a three dimensional (3D) grid with a maximum size of 20 × 20 × 20 Å. The grid box was placed at the centroid of the predicted active site area. A few important amino acid residues including Ser40, Cys47, His98, His104, Ser148 and Asp187 from molecules A and C were selected as the active site residues as proposed by Liao et al. (2001). The ligands were docked to the protein using the Glide docking protocol after the receptor grid was created.
2.4 Virtual screening workflow
The virtual screening workflow utilized the GLIDE module available in the Schrödinger suite to dock and rank lead-like compounds in order to classify the possible ligands. Initially, the compounds were filtered based on Lipinski’s rule of five followed by three different docking stages. The first stage performed HTVS (high throughput virtual screening) docking to estimate protein–ligand binding affinities. Then 10% of the best docked ligands were preserved and transferred to the next stage, which performed SP (standard precision) docking. At this stage, the 10% successful ligands were moved to the XP (extra precision) docking stage for further refinement of good ligand pose. Finally, the best poses were generated by Glide XP and ranked according to the Glide score (Friesner et al., 2006; Halgren et al., 2004). Tighter binders are represented by more negative Glide score values. The compounds with the highest ranks were considered for further analysis. In comparison with the most potent RS inhibitor reported, molecular docking of the LkRS with potential hits was also carried out with YASARA. The complex of compounds that bound with the receptor in a good docking position would demonstrate higher binding energy and lower Kd value, where positive binding energy meant stronger binding energy (Yadav et al., 2017).
2.5 In silico ADME and drug-likeness prediction
The prediction of the ADME properties of the potential hit compounds were assessed using the SwissADME webserver (Daina et al., 2017). In order to explore the appropriate pharmacokinetics profiles, a number of physicochemical and drug-like properties, including properties under the Rules of five, were recorded. The TPSA (<100), log P (<5) and molecular weight (<500) values in the ADMET studies indicated good oral bioavailability, whereas the number of rotatable bonds (<25) and the number of hydrogen bond acceptors (<10) or donors (<5) indicated good intestinal availability (Damale et al., 2019).
2.6 Molecular dynamics simulation
Molecular dynamics simulations were performed using YASARA Structure v.18.4.24 employing the AMBER14 force field (Krieger and Vriend, 2015). The simulations were run at 310 K with explicit water under periodic boundary conditions. The simulation cells were filled with water to a density of 0.997 g/cm3, and counter ions were added to a final concentration of 0.9% NaCl. Electrostatic interactions were treated with an 8 Å cut-off radius, using the Particle Mesh Ewald algorithm. To imitate the physiological conditions, the pH of the system was retained at 7.4. The simulation protocol began with the optimization of the hydrogen-bond network, consequently, a cubic simulation cell was created in the periodic boundary state, where the protein of each complex was parameterized. A total of four different systems were used to execute the MD simulations on the Windows operating system desktop PC (Intel® Core™ i7-6700 Processor @ 3.40 GHz) with 16.00 GB of RAM. The trajectory was computed for 60 ns.
3 Results and discussion
3.1 Homology modelling of riboflavin synthase from Leptospira kmetyi
The riboflavin synthase (Accession No. WP 010575348.1.) of Leptospira kmetyi is 201 amino acid residues in-length with a molecular weight of 22.3 kDa. The PSI-BLAST analysis showed that there were three crystal structures of low identity homologous riboflavin synthase available in the Protein Data Bank. LkRS shares the highest identity of 39% with the three-dimensional structure of Escherichia coli riboflavin synthase (PDB ID: 1I8D). The next two closest riboflavin synthase crystal structures are from Brucella abortus (PDB ID: 4EOF) and Schizosaccharomyces pombe (PDB ID: 1KZL) with 37% and 35% identities, respectively. Fig. 1 shows the multiple sequence alignments of the target sequence (LkRS) against three riboflavin synthases with known 3D structures. The catalytic triads (S148, H98, D187) and the binding sites (S40, C47, T150, H103) play central roles in protein function. Multiple sequence alignments display structurally and functionally important conserved regions within a protein family (Wiltgen, 2019). The substrate-binding MFTG motif situated in the N-terminus is important in stabilizing the active site architecture (Long et al., 2012; Illarionov et al., 2001; Liao et al., 2001). The YASARA software was used to model LkRS using the riboflavin synthase 3D structure from E. coli (EcRS) as a template. The structure was modelled as a homotrimer of 23 kDa subunits (Fig. 2(a)). The stereochemical quality of the modelled LkRS was assessed by using PROCHECK (Laskowski et al., 1993). The Ramachandran plot analysis of LkRS (Fig. 2(b)) showed that only one residue in the outlier region, 9.9% in the allowed region and 90% of the residues were found in the favored region. This indicated that 90% of the residues in the protein structure satisfied the phi and psi dihedral angle distributions. The ProSA-web server is commonly used for examining 3D protein structure models for possible errors (Wiederstein and Sippl, 2007). Fig. 2(c) shows that the residue energies of the LkRS model were negative for almost all the amino acid residues. The Z-score for the final LkRS model of −6.65 was in the range of the native conformations for crystal structures (Fig. 2(d)). These results indicated the acceptability of the predicted model. Z-scores are generated by the overall quality score calculated by ProSA-web, where problematic or erroneous parts of a model are positive in values. The nature of the trimer configuration suggests only one active site of riboflavin synthase at the interface of two neighboring monomers and the other two solvent exposed active sites are non-functional (Liao et al., 2001; Mishra et al., 2015). There are two of the triad Ser146 → His97 → Asp185 in chains A and C. The other two dyads are Ser41 and Cys48 (both in chain C), and dyads of Thr148 (chain A) and His102 (chain C). However, Serer et al. (2014), found only one active site at the interface between two subunits of Brucella abortus riboflavin synthase.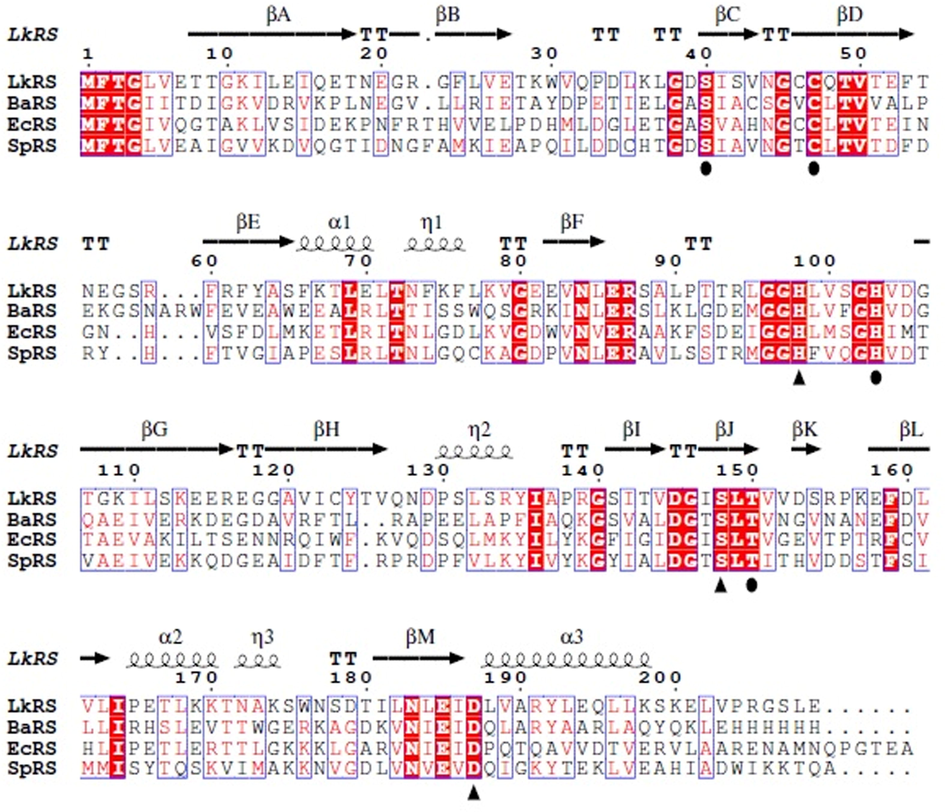
Multiple sequence alignments of LkRS and known 3D structures of riboflavin synthase from B. abortus (BaRS), E. coli (EcRS) and S. pombe (SpRS). The alignments were performed with Clustal Omega and visualized by ESPript. The predicted secondary structures of LkRS are shown above the alignment where helices are indicated by springs, strands by arrows and turns by TT letters. The conserved amino acids are colored in red. Triangle and circle symbols indicate the important amino acid residues for the catalytic triad motif and the binding site, respectively.
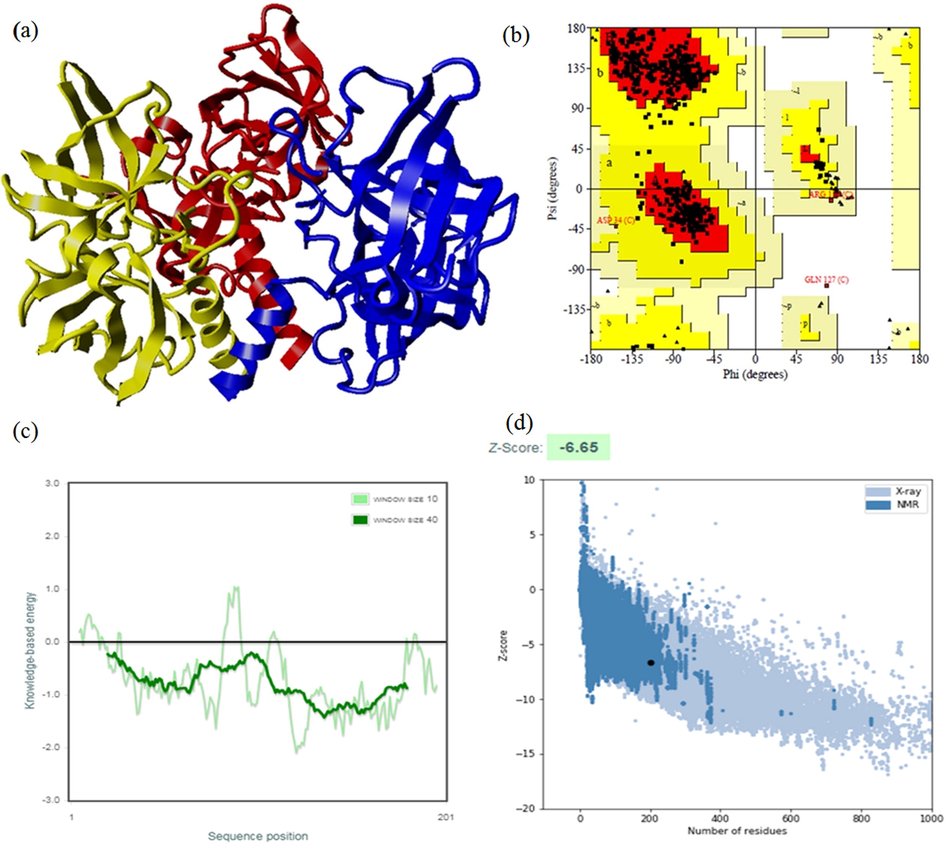
a) Modelled structure of homotrimeric riboflavin synthase coloured in red (chain A), blue (chain B), yellow (chain C) via YASARA. b) Ramachandran plot for predicted LKRS model. c) Energy plot for the predicted LKRS. d) ProSA-web Z-scores of all protein chains in PDB determined by X-ray crystallography (light blue) and NMR spectroscopy (dark blue) with respect to their length. The Z-score of LKRS was presented in the range depicted in black dot.
3.2 Molecular docking and binding pose analysis
The virtual screening technique plays a significant role in structure-based drug discovery. It shortens the drug discovery pipeline by screening large sets of inhibitors from a library in an economical way. As reported by Mishra et al. (Mishra et al., 2015), this approach was successfully applied in identifying 15 potential inhibitors of neuraminidase. In this study, a receptor grid was generated for docking analysis using the active sites extrapolated from the template structure EcRS. Top hit compounds (Table 1) were obtained through the virtual screening of one million compounds from the ZINC database with LkRS as a druggable target. Riboflavin synthase was used as a druggable target because it is the key enzyme catalyzing two molecules of 6,7-dimethyl-8-ribityllumazines to the precursor riboflavin and 5-amino-6-ribitylamino-2,4(1H,3H)-pyrimidinedione (Gerhardt et al., 2002; Ladenstein et al., 2013). Yang et al. (2011), proposed riboflavin synthase as an important immunogenic candidate for use as a vaccine against brucellosis. In this research, the promising compounds with the top three highest docking scores were ZINC21883831 (Compound 1), ZINC66132835 (Compund 2) and ZINC38739608 (Compound 3) with Glide scores of −10.987, −10.672 and −10.464 kcal/mol, respectively. The parameters of pharmacokinetics including absorption, distribution, metabolism and excretion (ADME), describe the molecule's biophysical or biochemical states during the route from intake to the target location. These parameters were predicted via the use of the SwissADME webserver (Daina et al., 2017). The Lipinski rule of 5, GI absorption, BBB permeant and cytochrome inhibition were determined from the analyses using this server. The results obtained increase the likelihood that these compounds have potential for use as drugs (Lipinski, 2004). According to Lipinski’s rule, a compound is considered orally bioactive if its physicochemical properties are within the acceptance limits. These criteria are molecular weight must be <500 Da, not more than 5 hydrogen bond donors, not more than 10 hydrogen bond acceptors and an octanol–water partition coefficient log P which must not exceed 5 (Lipinski, 2004). The physiochemical profiles and the ADME properties of the top three potential hits and the reported potent riboflavin synthase inhibitor are shown in Table 2. These compounds have molecular weights between 360 and 394 g/mol and comprise of 2H-bond donor groups, and 3H-bond acceptor groups except that the reported potent riboflavin synthase inhibitor has 7H-bond donors and 7 acceptor groups. These compounds recorded 59–184 Å2 of total polar surface area (Tpsa) with 4–7 rotatable bonds. Molecular weight is a significant factor for evaluating the permeability of a drug candidate. Compounds with higher molecular weights may not pass the blood brain barrier as easily as those with lower weights (Waring, 2009). The molecular weight, HBD and HBA of these compounds were within the appropriate ranges in accordance with Lipinski’s rule of five. These compounds would have worse permeabilities if their values were above the acceptable ranges. The TPSA value should be lower than 100 Å2 as this affected drug absorbance and distribution (Abad-Zapatero et al., 2014; Lipinski, 2004). The TPSA values for the potential drug compounds are in the acceptance range except for compound 3. However, there is another descriptor identified by Veber et al. (2002) where the acceptable range values of TPSA are lower than 140 Å2. According to Veber’s rule, a good oral bioavailability compound possesses 10 or fewer of rotatable bonds and the topological polar surface areas are not greater than 140 Å2 (Veber et al., 2002). However, the TPSA value for the reported potent riboflavin synthase inhibitor is 184.43 Å2. All of these compounds comply with the five Lipinski and Veber rules, indicating that these compounds have ideal oral bioavailabilities (Zerroug et al., 2019). From the virtual screening, these three hit compounds were selected as potential inhibitors of LkRS. As reported by Cushman et al. (2001), 9-D-ribityl-1,3,7-trihydropurine-2,6,8-trione (Compound 4) was identified as the most potent riboflavin synthase inhibitor particularly for E. coli. For the prediction of a more reliable outcome and for comparison with compound 4, all the three hit compounds along with compound 4 were therefore redocked into the same binding pocket via YASARA. The docked complexes were analyzed for comparative binding energies and dissociation constants (Kd). The findings suggested that the residues Ser40, Cys47, His98, His103 and Ser148 were the main residues that corresponded to the different kinds of interactions in the binding pockets. The docking results revealed better binding energies than that of Compound 4 (8.23 kcal/mol). Among the three possible hits, LkRS-Compound 1 exhibited the best binding energy of 10.13 kcal/mol. whereas the binding energies for LkRS-Compound 2 and LkRS-Compound 3 were 10.25 and 10.46 kcal/mol, respectively. Moreover, LkRS-Compound 1 had the lowest dissociation constant (Kd) value of 0.04 µM compared to LkRS-Compound 2 (0.03 µM), LkRS-Compound 3 (0.02 µM) and LkRS-Compound 4 (0.9 µM). The docking position of the LkRS complexed with each compound (compound 1–4) were superimposed using the LkRS-Compound 4 complex as a point for comparison. The superposition of LkRS-Compound 1 with LkRS-compound 4 showed an RMSD of 0.0939 Å. while the RMSD values for the superposed complex LkRS-Compound 2 and LkRs-Compound 3 were 0.1369 Å and 0.1137 Å, respectively. The superimposed views of the docked conformations are shown in Fig. 3. These results shed light on the potential of Compounds 1, 2 and 3 as new inhibitors for riboflavin synthase. HBD = Hydrogen bond donor, HBA = Hydrogen bond acceptor, TPSA = Total polar surface area, BBB = Blood-brain barrier.
Compound
ZINC ID/IUPAC Name
Docking Score (kcal/mol)
Glide score
(kcal/mol)
1
ZINC21883831
2-[(2-chloro-4-fluoro-phenyl)methylsulfanyl]-7-phenyl-3,5-dihydropyrrolo[2,3-e]pyrimidin-4-one−10.987
−10.987
2
ZINC66132835
2-[3-(2-phenoxyethoxy) phenyl]-2,3-dihydro-4(1H)-quinazolinone−10.672
−10.672
3
ZINC38739608
N-[4-[2-(1-naphthyl amino)-2-oxo-ethyl] thiazol-2-yl]thiophene-3-carboxamide−10.464
−10.464
4
ZINC38739665
N-[4-[2-[(4-benzyloxyphenyl) methylamino] −2-oxo-ethyl]thiazol-2-yl] thiophene-3-carboxamide−10.464
−10.464
5
ZINC06577075
2-[[4-(2,3-dimethylphenyl)piperazin-1-yl] carbonylmethyl]-4-(4-pyridylmethyl) phthalazin-1-one−10.401
−10.401
6
ZINC59420747
2-(3-bromo-4-fluoro-phenyl)-N-[(4R)-4-methyl-2,5-dioxo-4-phenethyl-imidazolidin-1-yl]acetamide−10.398
−10.398
7
ZINC78114635
3-fluoro-N-[(1S)-1-[4-(pyridine-4-carbonylamino)phenyl]ethyl]pyridine-4-carboxamide−10.349
−10.349
8
ZINC12894491
(S)-3-(1-(2-(1H-indol-3-yl)ethyl)-2,5-dioxoimidazolidin-4-yl)-N-(2-chlorobenzyl) propanamide−10.296
−10.296
9
ZINC57755724
(5R)-3-[(2R)-2-hydroxy-3-(1-naphthyloxy) propyl]-5-methyl-5-phenyl-imidazolidine-2,4-dione−10.280
−10.280
10
ZINC77781669
1-[(2S)-4-hydroxy-2-phenyl-butyl]-3-[[4-(1,2,4-triazol-1-yl)phenyl]methyl]urea−10.260
−10.260
Molecule
Compound 1
Compound 2
Compound 3
Compound 4
Formula
C19H13CIFN3OS
C22H2ON2O3
C20H15N3O2S2
C10H14N4O7
MW (g/mol)
385.84
360.41
393.48
302.24
Rotatable bonds
4
6
7
5
HBA
3
3
3
7
HBD
2
2
2
7
TPSA (Å2)
86.84
59.59
127.57
184.43
XLOGP3
4.61
3.35
3.86
−4.27
ESOL Log S
−5.47
−4.28
−4.80
0.99
ESOL Solubility (mg/ml)
1.31e−03
1.88e−02
6.27e−03
2.95e + 03
GI absorption
High
High
Low
Low
BBB permeant
No
Yes
No
No
P-gp substrate
No
Yes
No
No
CYP1A2 inhibitor
Yes
Yes
Yes
Yes
CYP2C19 inhibitor
Yes
Yes
Yes
No
CYP2C9 inhibitor
Yes
Yes
Yes
No
CYP2D6 inhibitor
Yes
Yes
Yes
No
CYP3A4 inhibitor
Yes
Yes
Yes
No
Log Kp (cm/s)
−5.38
−6.12
−5.96
−11.18
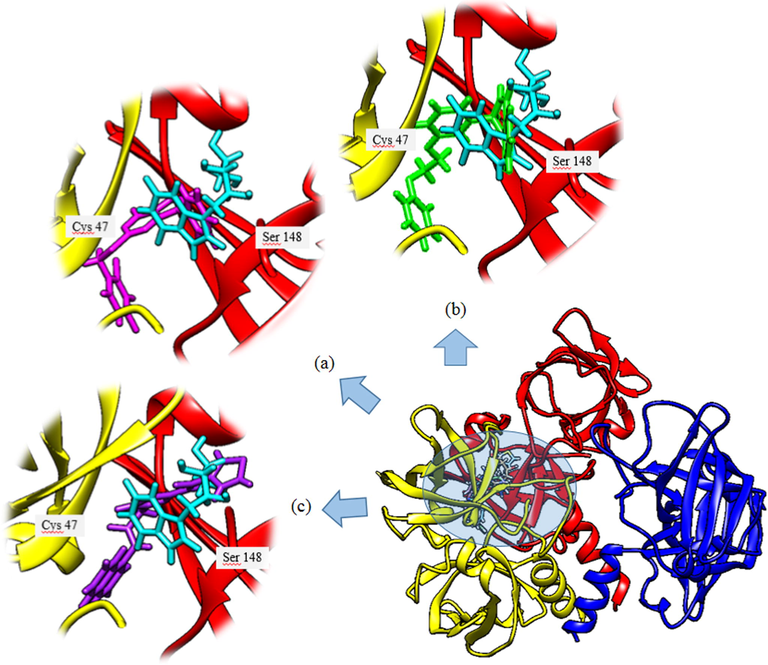
Three-dimensional (3D) representations of docked orientations of four different ligands bound to LkRS. The enlarged figure depicted the superimposed view of the docked conformation of compound 1 (magenta) (a), Compound 2 (green) (b), and Compound 3 (purple) (c) with Compound 4 (cyan) against LkRS.
3.3 Analysis of molecular dynamics simulation
MD simulation computes physical motions of biological molecules in particular environments. MD simulation helps in understanding the functional and structurally important behaviors of a large molecule in a real-time scenario. The docked complex of LkRS with the top three compounds (1, 2, and 3) were further evaluated using MD simulations to gain insights into the conformational stabilities and interactions of the receptor-ligand complexes in physiological environmental conditions. The MD simulation were performed to evaluate the dynamic behaviors of the prospective drug molecules against the druggable target of LkRS. Based on the RMSD analyses, the trajectories of all the complex systems were stable for 20–30 ns indicating that the systems had reached convergence and the simulated structures at 60 ns were comparatively stable. The RMSD measures the average distance between atoms to reveal structural changes during the simulation over time (Ishak et al., 2017). Fig. 4 shows the graph plots of RMSD between the apo structure of LkRS and the complex structure of LkRS with the top three potential leads model (LkRS-Compound 1, LkRS-Compound 2 and LkRS-Compound 3), respectively. LkRS-Compound 1 and LkRS-Compound 3 showed increments of RMSD values up to 6 Å. while the other two structures remained stable within the range of 3–4 Å after 10 ns. A higher RMSD value indicates the protein undergoes more structural changes probably due to the fitting of the ligand into the binding pocket by conformational rearrangements. Lower deviations are indicative of better stabilities (Damale et al., 2019). Towards the end of the simulation (60 ns), LkRS-Compound 3 showed the highest RMSD with a larger conformational change as compared to the other two complexed structures. This indicated the lower interaction efficiency of the compound with the riboflavin synthase. The mean RMSD values for the complexes of LkRS-Compound 1, LkRS-Compound 2 and LkRS-Compound 3 were 3.827, 2.916 and 3.539 Å, respectively. This suggested that the LkRS-Compound 2 complex had better stability than the other hit complexes. In addition, Ligplot plus version 4.5.3 (http://www.ebi.ac.uk/thornton-srv/software/LIGPLOT) was used for the 2D visualization of the ligand–receptor interaction. The LigPlot analysis enables us to understand the interaction pattern between the docked compounds and the protein residues especially hydrophobic interactions as well as the hydrogen bonding pattern (Wallace et al., 1996). Non-bonded interactions such as hydrogen bonds and hydrophobic interactions stabilize the protein–ligand complex structures (Salentin et al., 2014). Fig. 5 shows the 2D interaction diagrams for the complex of LkRS with the top 3 hit compounds after 60 ns simulation time, respectively. It showed that, Compound 1 bound between chain A and chain C by forming 3 hydrogen bonds with Val162(A), Ile164(A) and Thr167(A) together with eight hydrophobic interactions with Gly96(A), Leu149(A), Thr150(A), Leu161(A), Leu163(A), Ser40(C), Thr49(C) and Thr68 (C). Compound 2 formed two hydrogen bonds both with Cys47 from chain C and twelve hydrophobic interactions with Thr150(A), Val162(A), Ile164(A), Thr167(A), Cys46(C), Gln48(C), Thr49(C), Thr68(C), Leu71(C), Thr72(C), Gly102(C) and His103(C). Compound 3 formed only one hydrogen bonds with Ile64(A) and twelve hydrophobic interactions with Leu5(A), Gly96(A), Gly97(A), Arg139(A), Thr150(A), Val162(A), Leu163(A), Ser40(C), Cys47(C), Thr68(C), Leu71(C) and His103(C). Table 3 shows the residues involved in the interactions of hydrogen bonds and the hydrophobic interactions between the compounds and the LkRS, respectively. The flexibility of each residue was assessed by its root mean square fluctuation (RMSF). The average RMSF value for each residue was determined from the trajectories in order to obtain information on the local structural flexibility. The RMSF values of the four systems were computed to analyze the flexibility and rigidity of the individual residues in the absence and presence of the ligands. Fig. 6 shows the RMSF values of the four systems calculated from 60 ns trajectories. Higher RMSF values indicate greater flexibility during the MD simulation. For all the complex structures of LkRS, greater RMSF changes were spotted in the C-terminal region of Chain B, specifically between Lys 198 to Glu 201 with RMSF values up to 7 Å. Moreover, RMSF changes was also seen in the C-terminal region of Chain C. The most flexible termini (N- or C-terminus) affect protein folding and also native state stability (Amini-Bayat et al., 2012; Krishna and Englander, 2005). The other regions showing higher fluctuations were Trp 30 for chain B and Glu 117 for chain C located in the loop regions, which were more flexible than β-sheets and α-helices. The fluctuations encountered in this region correlated with the high flexibility nature of the loops. The overall magnitude of the fluctuations was the highest for the complex structure of LkRS-Compound 3 which explained the weak binding efficiency of this compound compared to the others. The radius of gyration (Rg) and the solvent accessible surface area (SASA) of the receptor-ligand complexes were also analyzed to investigate the compactness of the structure and the transition of the buried residues as a result of ligand binding, respectively. Kamaraj and Purohit (2013) defined Rg as the mass-weight root-mean-square distance of the collection of atoms from their common center of mass. Rg is a significant indicator in predicting the structural activity of macromolecules. It provides information on the overall dimension of the protein. Changes in Rg were due to conformational changes as the lead compound bound to the protein (Seeliger and Groot, 2010). The complex structure of LkRS-Compound 3 showed a higher Rg value compared to the others (Fig. 7(a). This might be due to the less tight packing with this compound. The structure of the highest ranked compound, LkRS-Compound 1 showed the lowest Rg value indicating the compound was tightly bound to the protein. The compactness of the ligand–protein complex is lower when the Rg value is higher, causing the interactions between the ligand and the protein to be weaker signifying a less stably folded protein (Liao et al., 2014; Dhankhar et al., 2020). Comprehension of the Rg provides information on the structural compactness and binding pattern of protein–ligand complex. Solvent-accessible surface area (SASA) tests the free energy of non-polar solvents when exposed to solvent. The binding affinity of protein–ligand interactions is correlated with the dissolving effects of protein, ligand, and rearrangement of molecules in solvents. The differences in the SASAs between all the complexes of the structures indicated the rearrangements of the respective trimeric structures and ligands. The SASA values for all four systems during MD were relatively stable especially at the ends of the simulations (Fig. 7(b)). According to the results, LkRS-Compound 1 showed less accessibility and interaction with the solvent that might be due to the tight binding of the compound when compared to the others.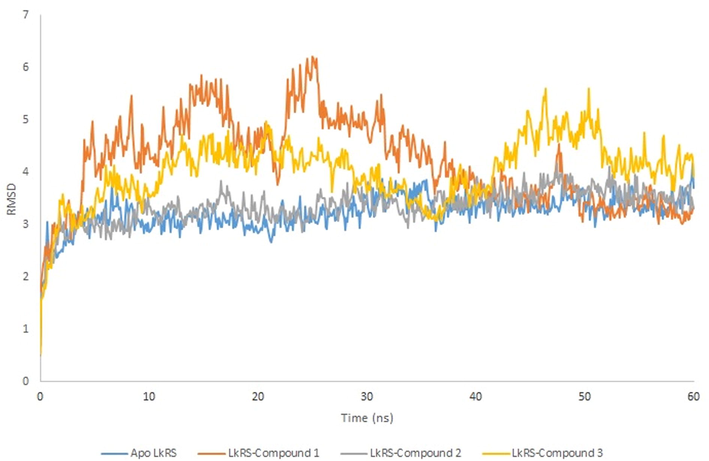
RMSD of the Cα-backbone atoms of riboflavin synthase complexes as a function of time. Apo structures and complexes were represented in blue, orange, grey and yellow, respectively.
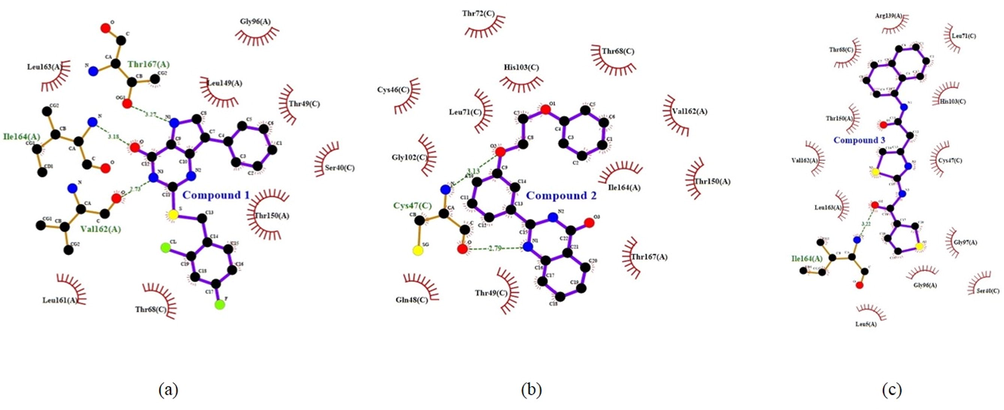
The interactions between the top three compounds; (a) Compound 1 (ZINC21883831) (b) Compound 2 (ZINC66132835) and (c) Compound 3 (ZINC38739608) with riboflavin synthase after 60 ns MD simulation. Schematic 2D representations of ligand–protein interactions were generated via LigPlot. Interactions seen are those mediated by hydrogen bonds and hydrophobic contacts. Hydrogen bonds are indicated by dashed lines between the atoms involved, while hydrophobic contacts are represented by an arc with spokes radiating towards the ligand atoms they come into contact with. The atoms in touch are shown with spokes radiating out.
Compound
Hydrogen bond
Hydrophobic
1
Val162 (A)
Ile164 (A)
Thr167 (A)Gly96 (A), Leu149 (A), Thr150 (A), Leu161 (A), Leu163 (A), Ser40 (C), Thr49 (C), Thr68 (C)
2
Cys47 (C)
Cys47 (C)Thr150 (A), Val162 (A), Ile164 (A), Thr167 (A), Cys46 (C), Gln48 (C), Thr49 (C), Thr68 (C), Leu71 (C), Thr72 (C), Gly102 (C), His103(C)
3
Ile164 (A)
Leu5 (A), Gly96 (A), Gly97 (A), Arg139 (A), Thr150 (A), Val162 (A), Leu163 (A), Ser40 (C), Cys47 (C), Thr68 (C), dLeu71 (C), His103 (C)
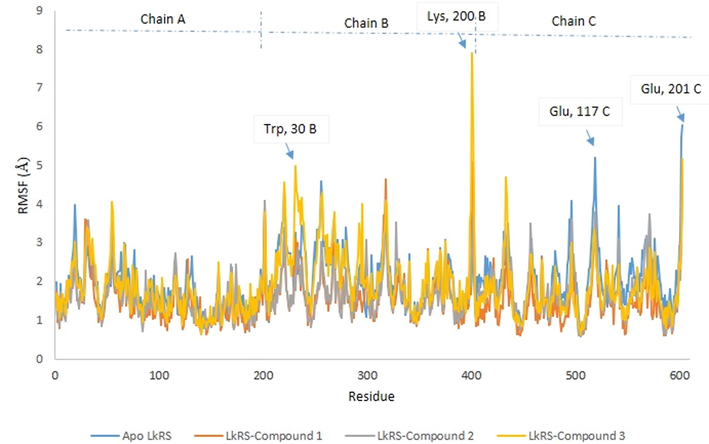
Profile of Root Mean Square Fluctuation (RMSF) of apo- and complex structure of LkRS.
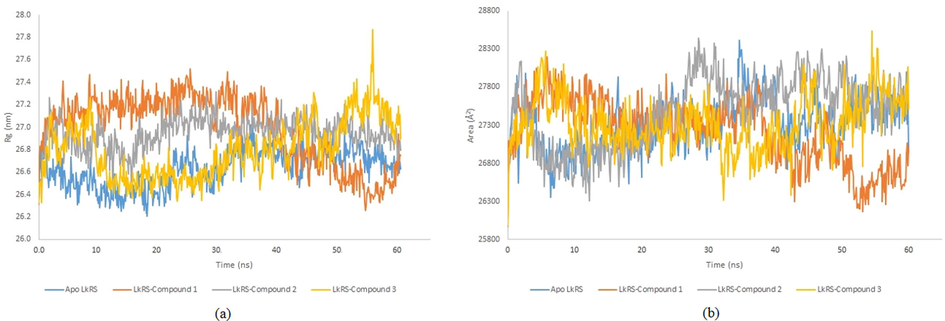
Profile of (a) Radius of gyration and (b) Solvent accessible surface area (SASA) of apo- and complex structure of LkRS respectively.
4 Conclusion
This study utilized the bioinformatics approach aimed at identifying potential inhibitors of riboflavin synthase from Leptospira kmetyi (LkRS). The three-dimensional structure of LkRS was predicted by homology modelling and was confirmed as being a good quality model via PROCHECK and ProSA-web validation with Z-score value of −6.65. Virtual screening was employed to classify drug compounds that were predicted to have high binding affinities for LkRS. ZINC21883831 (Compound 1), ZINC66132835 (Compound 2) and ZINC38739608 (Compound 3) were the top three highest ranked Glide scorers with ratings of −10.987, −10.672 and −10.464 (Kcal/mol), respectively from over a million screened compounds. In addition, all of the pharmacokinetic properties expected were within the relevant limits. To gain deeper insights into the driving forces between the inhibitors and LKRS, molecular dynamics simulation of LkRS with the top hit compound was performed. Based on these in silico results, ZINC21883831 (Compound 1), ZINC66132835 (Compound 2) and ZINC38739608 (Compound 3) have the potential to be developed as antibacterial compounds targeting LkRS. The compound with the greatest potential for inhibiting LkRS is ZINC21883831. Insights from this research shed light on the development of new potential antibacterial drugs that may be beneficial in the treatment of leptospirosis or other related infectious diseases. Further experimental studies in vitro and in vivo are being considered to further validate these results.
Acknowledgement
The study was supported by a grant from the Ministry of Science, Technology & Innovation, Malaysia. (Sciencefund: 02-01-04-SF1321).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Alternative variables in drug discovery: promises and challenges. Future Med. Chem. Engl.. 2014;6:577-593.
- [Google Scholar]
- Relationship between stability and flexibility in the most flexible region of Photinus pyralis luciferase. Biochim. Biophys. Acta. 2012;1824:50-58.
- [Google Scholar]
- Overlapping riboflavin supply pathways in bacteria. Crit. Rev. Microbiol.. 2017;43(2):196-209.
- [Google Scholar]
- An Atypical Riboflavin Pathway Is Essential for Brucella abortus Virulence. PLoS ONE. 2010;5(2)
- [Google Scholar]
- Serovar diversity of pathogenic Leptospira circulating in the French West Indies. PLoS Negl Trop. Dis. 2013
- [Google Scholar]
- What has computer-aided molecular design never done for drug discovery. Expert Opin. Drug Discov.. 2006;1:103-110.
- [Google Scholar]
- Global morbidity and mortality of leptospirosis: a systematic review. PLoS Neg. Trop. Dis. 2015
- [Google Scholar]
- Design, synthesis, and evaluation of 9-D-ribityl-1,3,7-trihydro-2,6,8-purinetrione, a potent inhibitor of riboflavin synthase and lumazine synthase. J. Org. Chem.. 2001;66(25):8320-8327.
- [Google Scholar]
- SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.. 2017;7:42717.
- [Google Scholar]
- Molecular docking, pharmacophore based virtual screening and molecular dynamics studies towards the identification of potential leads for the management of H. pylori†. R. Soc. Chem. Adv.. 2019;9:26176-26208.
- [Google Scholar]
- In-silico approach to identify novel potent inhibitors against GraR of S. aureus. Front. Biosci., Landmark. 2020;25:1337-1360.
- [Google Scholar]
- Troubleshooting the heterologous expression of riboflavin synthase from Photobacterium sp. J15. Eur. J. Biomed. Pharm. Sci.. 2016;3:699-705.
- [Google Scholar]
- Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein–ligand complexes. J. Med. Chem.. 2006;49:6177-6196.
- [Google Scholar]
- Studies on the reaction mechanism of Riboflavin synthase: X-ray crystal structure of a complex with 6-Carboxyethyl-7-Oxo-8-ribityllumazine. Structures. 2002;10:1371-1381.
- [Google Scholar]
- The Light and dark sides of virtual screening: what is there to know. Int. J. Mol. Sci.. 2019;20(6):1375.
- [Google Scholar]
- Guidelines for the diagnosis, management, prevention and control of Leptospirosis in Malaysia. Ministry of Health Malaysia; 2011.
- Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem.. 2004;47(7):1750-1759.
- [Google Scholar]
- In-silico studies of some Oxadiazoles derivatives as anti-diabetic compounds. J. King Saud Univ. Sci.. 2020;32:423-432.
- [Google Scholar]
- Riboflavin synthase of Escherichia coli Effect of single amino acid substitutions on reaction rate and ligand binding properties. J. Biol. Chem.. 2001;276(15):11524-11530.
- [Google Scholar]
- Molecular dynamic simulation of space and earth-grown crystal structures of thermostable T1 lipase Geobacillus zalihae revealed a better structure. Molecules. 2017;22(10):1574.
- [Google Scholar]
- In Silico Screening and Molecular Dynamics Simulation of Disease-Associated nsSNP in TYRP1 Gene and Its Structural Consequences in OCA3. BioMed Res. Int. 2013
- [Google Scholar]
- Increasing the precision of comparative models with YASARA NOVA—a self-parameterizing force field. Proteins. 2002;47:393-402.
- [Google Scholar]
- New ways to boost molecular dynamics simulations. J. Comput. Chem.. 2015;36:996-1007.
- [Google Scholar]
- The N-terminal to C-terminal motif in protein folding and function. Proc. Natl. Acad. Sci. U.S.A.. 2005;102:1053-1058.
- [Google Scholar]
- The lumazine synthase/Riboflavin synthase complex: shapes and functions of a highly variable enzyme system. Fed. Eur. Biochem. Soc. J.. 2013;280:2637-2663.
- [Google Scholar]
- PDBSum more: new summaries and analysis of the known 3D structure of proteins and nucleic acids. Nucleic Acids Res.. 2005;33:266-268.
- [Google Scholar]
- PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr.. 1993;26:283-291.
- [Google Scholar]
- Ligand-based and structure-based investigation for Alzheimer’s disease from traditional chinese medicine. Evidence-Based Complementary Altern. Med. 2014
- [Google Scholar]
- Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol.. 2004;1:337-341.
- [Google Scholar]
- Molecular dynamic simulations and novel drug discovery. Exp. Opin Drug Dis.. 2017;13
- [Google Scholar]
- Riboflavin biosynthesis and regulatory factors as potential novel anti-infective drug targets. Chem. Biol. Drug Des.. 2012;75:339-347.
- [Google Scholar]
- Ligand based virtual screening for identifying potent inhibitors against viral neuraminidase: an in silico approach. J. Taibah Univ. Sci.. 2015;9:20-26.
- [Google Scholar]
- Binding of the anticancer drug zd1694 to E. Coli thymidylate synthase: assessing specificity and affinity. Structures. 1996;4:1317-1324.
- [Google Scholar]
- Polypharmacology rescored: Protein-ligand interaction profiles for remote binding site similarity assessment. Prog. Biophys. Mol. Biol.. 2014;116:174-186.
- [Google Scholar]
- Conformational transitions upon ligand binding: Holo-structure prediction from apo conformations. PLoS Comput. Biol.. 2010;6(1)
- [Google Scholar]
- Crystallographic and kinetic study of Riboflavin synthase from Brucella abortus, a chemotherapeutic target with an enhanced intrinsic flexibility. Acta Cryst. Sect. D Biol. Cryst.. 2014;D70:1419-1434.
- [Google Scholar]
- Leptospira kmetyi sp. nov. isolated from an environmental source in Malaysia. Int. J. Syst. Evol. Microbiol.. 2009;59:705-708.
- [Google Scholar]
- Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem.. 2002;45:2615-2623.
- [Google Scholar]
- LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Prot. Eng.. 1996;8:127-134.
- [Google Scholar]
- Defining optimum lipophilicity and molecular weight ranges for drug candidates molecular weight dependent lower logD limits based on permeability. Bioorg. Med. Chem. Lett.. 2009;19:2844-2851.
- [Google Scholar]
- ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res.. 2007;35:W407-W410.
- [Google Scholar]
- Algorithms for structure comparison and analysis: homology modelling of proteins. Encycl. Bioinf. Comput. Biol. 2019
- [Google Scholar]
- Molecular docking studies of 3-bromopyruvate and its derivatives to metabolic regulatory enzymes: implication in designing of novel anticancer therapeutic strategies. In: Maga G., ed. PLoS One. 2017.
- [Google Scholar]
- Immunoproteomic analysis of Brucella melitensis and identification of a new immunogenic candidate protein for the development of brucellosis subunit vaccine. Mol. Immunol.. 2011;49:175-184.
- [Google Scholar]
- Virtual screening in drug-likeness and structure/activity relationship of pyridazine derivatives as Anti-Alzheimer drugs. J. King Saud Univ. Sci.. 2019;31(4):595-601.
- [Google Scholar]
- Evolution of vitamin B2 biosynthesis: 6,7-dimethyl-8-ribityllumazine synthases of Brucella. J. Bacteriol.. 2006;188:6135-6142.
- [Google Scholar]




