

Translate this page into:
Identification of phytochemical compounds to inhibit the matrix-like linker protein VP26 to block the assembles of white spot syndrome virus (WSSV) envelope and nucleocapsid protein of marine shrimp: In silico approach
⁎Corresponding author at: Ecological Physiology, King Abdulaziz University, Faculty of Science, Biological Sciences Department, P.O. Box 80203, Jeddah 21589, Saudi Arabia. moaljahdali@kau.edu.sa (Mohammed Othman Aljahdali)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
White spot syndrome virus (WSSV) is an enveloped pathogenic virus of cultured shrimp that has a mortality rate of up to 100 % in intensive aquaculture systems. The virulent pathogen causes an economic loss estimated at US$1 billion annually to global shrimp and prawn production. To date, no effective antiviral or vaccine has been progress that can reduce the economic losses caused by the pathogen. The complex genomic structure of the nucleocapsid of a virus, consisting of double-stranded DNA as genetic material surrounded by lipid envelope protein. It has been found that VP26, a major envelope protein interacts with the virus VP51 capsid and makes a bridge between the envelope and nucleocapsid protein resulting in mature virion of the virus. Blocking the interaction by targeting VP26 can hinder the mature virion production and non-infectivity of the virus. Therefore, in order to blocking the function of VP26, an in-silico drug design approach has been utilized to screen the potential antiviral compounds from the medicinal plant, Withania somnifera (WS) of Saudi Arabia. Isolated thirty-nine (39) phytochemicals compounds from the plant have initially been screened by molecular docking method. The best three compounds, namely, withanolide (CID: 118701104), Coagulin Q (CID: 10100411), and withanolide D (CID: 23266161) have been selected based on their docking score −8.2 kcal/mol, −8.1 kcal/mol and −7.9 kcal/mol, respectively, which have been further evaluated based on pharmacokinetics including ADME (Absorption, Distribution, Metabolism and Excretion) and toxicity approaches. Additionally, 100 ns molecular dynamics (MD) simulation methods confirmed the structural stability of the compounds to the binding sites of the VP26 protein. Therefore, the selected compounds can be utilized as an antiviral drug to fight against the WSSV virus, which will bring remarkable advancement in the aquaculture sector.
Keywords
In-silico drug design
VP26
Virtual screening
W. somnifera
Molecular dynamics simulation

1 Introduction
Shrimp culture have been increased day by day due to their high demand, which included a rich source of protein and omega-3 fatty acid as well as antioxidant astaxanthin (6, 11, 12, 13) (Karnila et al., 2020). However, the devastating effect of numerous virus disease has brought tremendous negative impacts on the shrimp industry. Among them, white spot syndrome virus (WSSV) is one that have been destroyed the shrimp industry since last three decades. Therefore, it led to great economical losses in every year such as about USD 6 million in 2016 by 100 % mortality within a week at many farms (Trang et al., 2019). Only genetic improvement fry namely specific pathogen free (SPF) fry has been established to promote sustainable shrimp farming.
White spot syndrome virus (WSSV) has been spreading white spot syndrome disease which is a highly contagious viral disease for farm based penaeid shrimps as well as wild shrimp (Cavalli et al., 2011). It mainly affects the gills and hepatopancreas of the shrimp, and damaged severely. WSSV is a doublestranded DNA virus (300 kbp) in the Nimaviridae family of the genus Whispovirus. WSSV has 22 capsid proteins that express four most abundant ones such as VP26, VP28, VP24 and VP19 where VP26 and VP28 constituted 60 % of the envelope protein (Zhou et al., 2009). There has a little knowledge about the predicted protein ∼184WSSV because there have no known homology sequences in the repositories. However, in the coding region of WSSV, there have some variations including two genomic deletions between ORFs wsv461/wsv464 (14/15) and ORFs wsv77/wsv502 (23/24), and a variable number of tandem repeats (VNTRs) occurring within wsv129 (ORF75), wsv178 (ORF94) and wsv249 (ORF125) (Liao et al., 2021). The variable regions are not only use for molecular marker but also have some relationships with viral evolution and infection phenotype which is apparent by the externally white sign of shrimp.
In GenBank, V26 has no similarity protein but contains 204 amino acids along with the estimated molecular mass of 22 kDa (Aljahdali et al., 2021a). The N terminal of VP26 is not only hydrophobic but also included a putative transmembrane anchor composed of a helix formed through the amino acid 12 to 34. The two α-amino acid have positive charge region behind the anchor with including C-terminal end of the protein from the cytoplasmic side. The cysteine is placed in the C-terminal domain indicate that the disulfide bond are not formed. It forms complex protein by interacting with other low abundance envelop protein, which occur naturally from VP26 (Robinson and Bulleid, 2020). It has a tegument feature and interacting with others proteins that may help to transport WSSV nucleocapsid to the host through the cytoskeleton. Therefore, V26 has been interacting with variety of protein molecules that plays a key role to virus invasion in later stage.
In the field of drug discovery, phytochemical bioactive compounds have a potential involvement in many cases to inhibit the disease related protein (Ling et al., 2014). These have been used because of easy availability, reasonable cost, and not harmful at all as well as the feasibility of oral administration. Withania somnifera (WS) has the anti-tumorigenic properties that act against the cancerous cell. It has also potential inhibitor for the treatment of neurodegenerative diseases. Moreover, In-silico drug design techniques is the best key for saving both the time and cost by predicting approximate protein and selecting small bioactive compounds (Aljahdali et al., 2021b). However, the viable phytochemical compounds are the critical objective for the drug design by CAAD. Therefore, molecular docking-based scoring function selected by a specific target, and the dynamic simulation confirm the stability of a drug candidate to the target receptor (Hasan et al., 2022). As a result, in-silico approaches of drug design will predict the new drug candidate against the specific virus by targeting suitable protein.
2 Materials and methods
2.1 Protein preparation
The structure of V26 protein has been taken from the large RCSB data bank (PDB). The WSSV protein V26 having a PDB identity 2EDM with molecular weight of 22 kDa was used. The protein was recovered and processed using AutoDockTools, which computed gasteiger charges, merged nonpolar hydrogen, added hydrogen atoms, and eliminated metal ions and cofactors (Aljahdali et al., 2021b).
2.2 Compounds retrieval and preparation
The bioactive compounds have been derived from the medicinal plant for drug design and discovery. Therefore, the popular database (https://cb.imsc.res.in/imppat/home) of Indian Medicinal Plant, Phytochemistry and Therapeutics (IMPPAT) has been surfing for the plant Withania somnifera. The database consisting of > 1742 Indian medical plants and about > 9500 phytochemical compounds which were utilized for natural based drug discovery.
2.3 Grid generation and active site identification
Active site (AS) of VP26 protein has been documented from the specific shape of protein through the CASTp 3.0 web server (https://sts.bioe.uic.edu/). The chemical reaction of protein was binding with a specific molecular substrate. Auto Dock vina, a virtual screening tool developed by PyRx, has been used to determine the receptor grid generation process and binding site from the complex AS (Samad et al., 2020).
2.4 Molecular docking
The PyRx virtual screening tool AutoDock Vina has been used for the molecular docking to detect the binding mode from the chosen receptor and designated phytochemical compounds. Entirely, the BIOVIS Discovery Studio Visualizer Tools 16.1.0.41 has been used for the observation of binding interaction of protein-ligands complex (Aljahdali et al., 2021b).
2.5 PK properties prediction
The drug absorption, distribution, metabolism, and excretion (ADME) mainly involved in drug to enter and out of the body are which related to the intensity and time course. The popular server SwissADME has been used in the study to evaluate the PK properties that can predict PK and drug-likeness properties of small molecules (Daina et al., 2017).
2.6 Toxicity prediction
Toxicity is the fundamental obstacle to drug design. The AdmetSAR 2.0 (https://lmmd.ecust.edu.cn/admetsar2/) web-based server was utilized to document toxicity from the selected compound.
2.7 Molecular dynamic simulations (MD)
The snapshots were taken from molecular dynamic simulation using Schrodinger's Maestro interface version v9.5. Desmond module in the Schrodinger package from Simulation Interaction Diagram (SID) were used for the analysis of simulation even as well as quality of molecular dynamic simulation (Aljahdali et al., 2022).
2.8 RMSD and RMSF analysis
RMSD was used for the estimate of average distance produced through the movement of a particular atom at the specific time compared to the reference time in MD simulation. In the study, time frames were aligned and estimated based on reference time (100 ns) from the RMSD of the protein fit ligand atom. On the other hand, the large number of residues generated by MD simulation, RMSF was employed as the preferred method for observing the conformational changes that occurred within a protein structure.
3 Results
3.1 Phytochemical retrieval and preparation
Initially, Indian natural and medicinal phytochemical compound library (IMPPAT) has been searched for the compounds from the desirable mangrove plant. Thirty-nine compounds has been identified from the mangrove plant Table (S1). The bioactive compounds have not only been retrieved but also saved in a 2D (SDF) file format. Optimized and prepared compounds were converted into pdbqt file format during the ligand preparation steps for further evaluation.
3.2 Active site identification and receptor grid generation
The popular web serve CASTp 3.0 was surfing to detect the binding site of protein from the separate AA restudies in a specific region. The complex AS pocket was identified from the retrieved binding site position of VP26 protein. The AS pocket were represented in the different colours by ball shape with 9 AA restudies (PRO102, THR104, PRO106, ASN115, MET116, THR117, TYR153, ASP170, CYS172) (Fig. 1).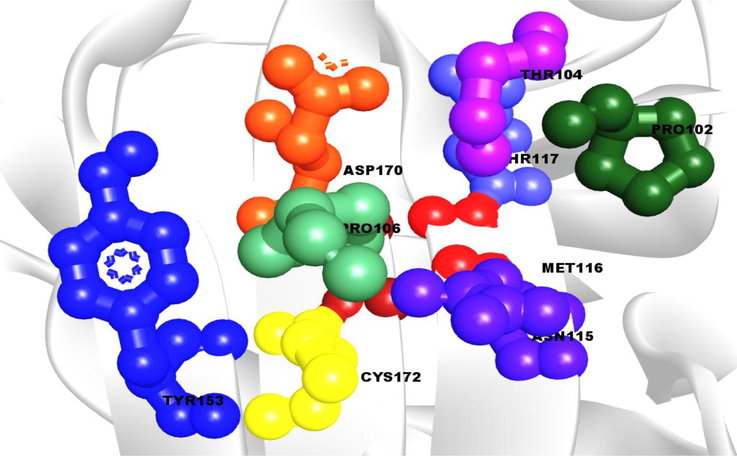
The active site and corresponding binding site of the VP26 protein are shown. The active sites (AS) of the VP26 protein are represented by a ball with red, green, yellow, and blue colors, which correspond to their binding site positions.
3.3 Molecular docking analysis
The thirty-nine phytochemical compounds have been selected from the marine plant of W. somnifera by using PyRx tools AutoDock vina wizard. The highest binding affinities range were −8.2 kcal/mol that found in alpha Amyrin. The distribution range of binding affinities between −8.2 and −2.5 kcal/mol were observed from the plant. The best three compounds have been selected by the binding affinities for in silico drug design. The additional table included information on the compound identities, chemical names, and 2D structures of the top three ligands as well as the binding affinities of the controls (S1).
3.4 Interpretations of protein–ligands interaction
The BIOVIA discovery studio visualizer was performed for observing the protein ligand interactions from the selected compounds and protein VP26 based on best binding affinities. The initially compound (CID118701104) was observed and were there had strong hydrogen bond, hydrophobic and Pi-Alkyl bonds. The hydrogen bond was formed at the position of ARG51, TYR46, ARG67 and MET49, and hydrophobic bond at the position of TYR69, MET50, THR71, GLN48 and PRO72 (Fig. 2).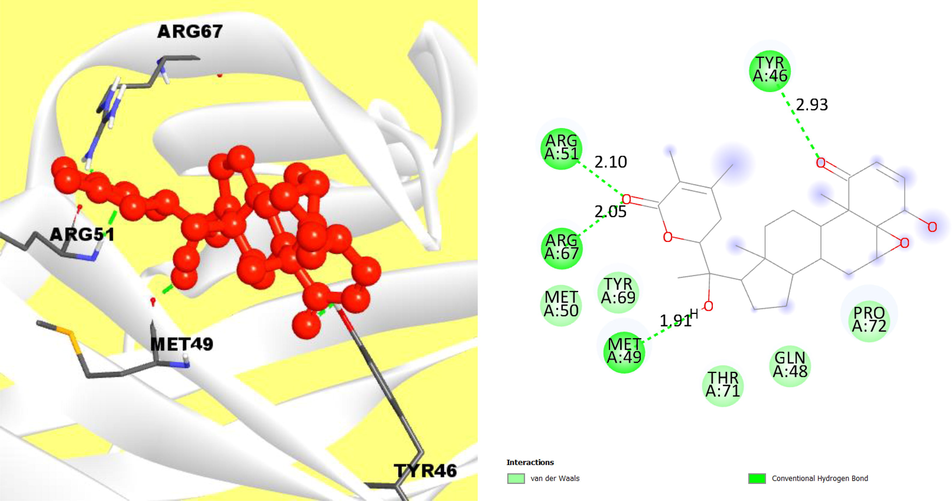
The interaction of protein and ligand between compound 118701104 (PubChem CID) and VP26 protein and left side representing 2D complex protein–ligand interaction.
Again, for the compound CID 11100411, it was clearly observed that it has one conventional hydrogen bond at the position of ARG136, ASN186, ASP132, PRO72 and TYR46 where there were hydrophobic bonds from the position of THR134, GLY188 and MET49 (Fig. 3).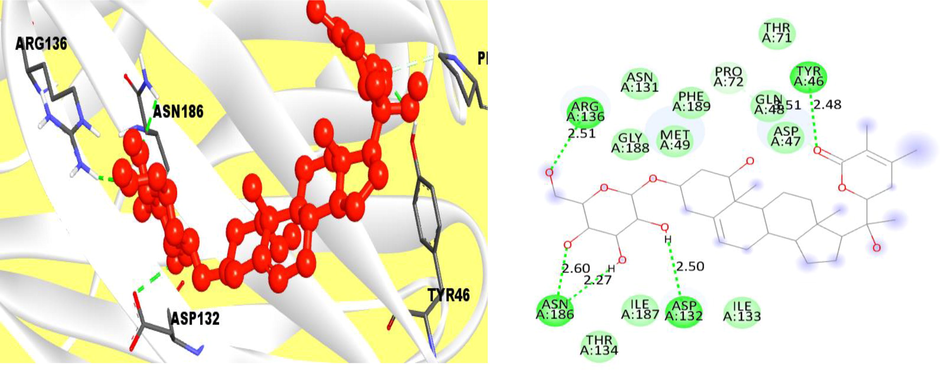
The interaction of protein and ligand between compound 11100411 (PubChem CID) and VP26 protein, and left side is representing 2D complex protein–ligand interaction.
In case of the compound CID23266161, a conventional strong hydrogen bond and carbon hydrogen bond were found between complex protein–ligand interaction. The conventional hydrogen bond at the position of MET49, GLN48, THR71, and PRO72 were found where the hydrophobic bond was located at the TYR46, ASP47, PRO72 and ASN70 position (Fig. 4).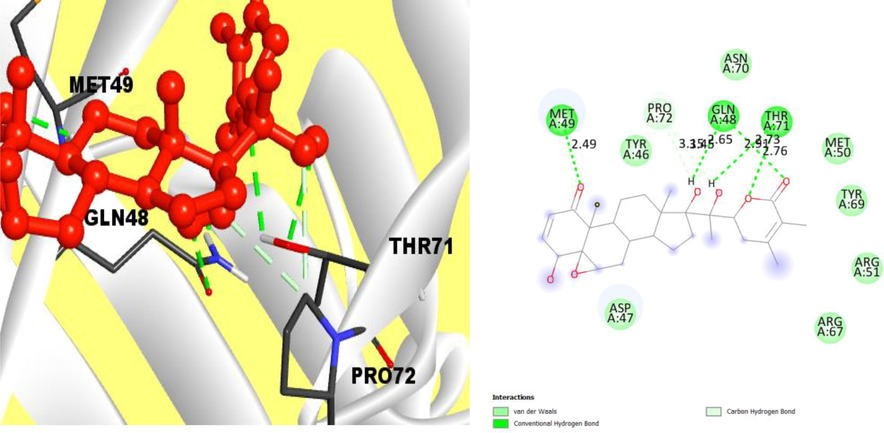
Interaction between the compounds CID: 23266161 and V26 protein. Left side indicates 3D interaction.
3.5 AMDE properties
The ADME analysed not only assist the physiochemical properties but also delivered the hypothesis for choosing top treatment candidates. Therefore, the pharmacological efficacy of the three selected compounds of W. somnifera has been demonstrated through the use of pharmacophore features such as physicochemical qualities, lipophilicity, water solubility, drug-likeness, and medicinal chemistry W. somnifera (Table 1).
Name
Properties
CID: 118701104
CID: 10100411
CID: 23266161
physicochemical properties
MW (g/mol)
470.60
620.77
486.60
Heavy atoms
34
44
35
Rotable bond
2
5
2
H-bond acceptors
6
10
7
H-bond donors
2
6
3
Lipophilicity
Log Po/w
3.74
3.76
3.42
Water solubility
Log S (ESOL)
−4.59
−4.88
−3.79
Pharmacokinetics
GI absorption
High
Medium
High
Drug-likeness
Lipinski
Yes
Yes
Yes
Medi. Chemistry
Synth. accessibility
Easy
Easy
Easy
3.6 Toxicity prediction
In silico drug design methods can reduce the large number of biological experimental test and eliminate toxic chemicals compounds in a short period. Therefore, admetSAR 2.0 web server has been used for identifying the toxicity of compounds. It has identified the hepatotoxicity, carcinogenicity, immunotoxicity, mutagenicity, and cytotoxicity through the admetSAR 2.0 web server (Table 2).
PubChem ID
Hepatotoxicity
Carcinogenicity
Immunotoxicity
Mutagenicity
Cytotoxicity
CID: 118701104
Inactive
No
Inactive
Inactive
Inactive
CID: 10100411
Inactive
No
Lightly active
Inactive
Inactive
CID: 23266161
Inactive
No
Inactive
Inactive
Inactive
3.7 MD simulation analysis
In the artificial environment, MD simulation has confirmed the stability of protein–ligand complexes. However, it has analysed the steady nature and conformation by performing MD simulation with 100 ns that observed the stability of the protein - ligand complexes. As a result, simulation trajectories have examined the RMSD, RMSF, intermolecular hydrogen bonds and protein - ligand contact analysis for the possible drug candidate.
3.8 RMSD analysis
The observed value and estimated value have changed with frame to a reference time to 1–3 Å or 0.1–0.3 nm that were entirely admissible. The protein structure (Cα) residues and ligand fit protein were analysed within 100 ns frame in the study. Therefore, VP26 protein from the WSSV and ligand from the compounds were estimated for the considerable fluctuation. All of compounds showed the optimum fluctuation > 3.0 Å. However, the fluctuations abnormality happened when the system was not properly equilibrated due to their requirement (Fig. 5).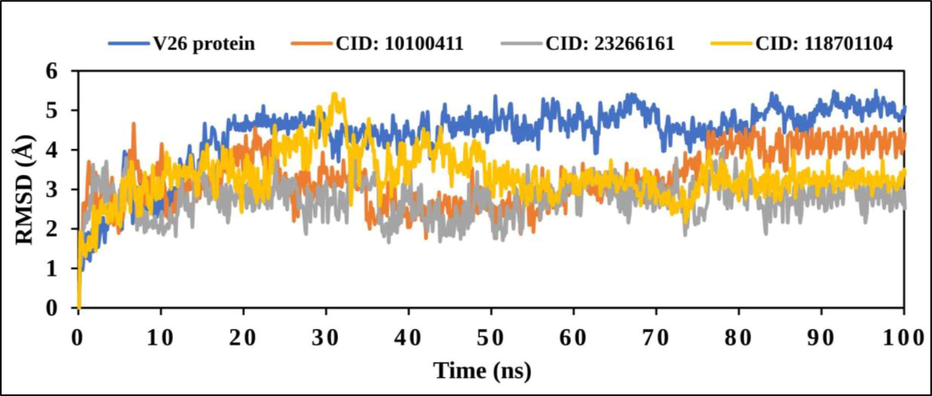
The graph represents the RMSD values from Cα atoms (blue curves) of VP26 protein and natural compounds, where the compounds have been shown as CID: 10100411 (red), CID: 23266461 (gray) and CID: 118701104 (yellow) with regards of 100 ns simulation time.
3.9 RMSF analysis
RMSF (root mean square fluctuation) was determined the positional difference between the protein and ligands in the particular time frame. It provided the RMSD value, evidence about protein heterogeneity and the movement of macromolecules through the performing data about RMSF. However, RMSF value provided the change of receptor along with amino acid chain for characterizing the protein (Fig. 6).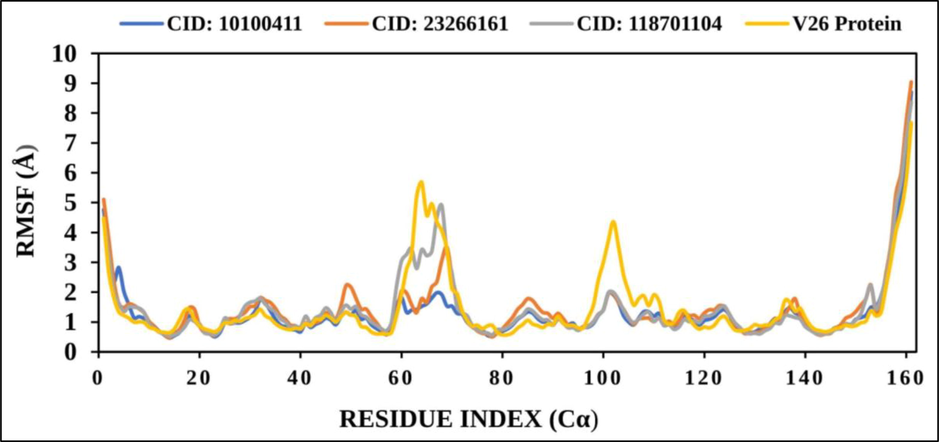
The line graph depicts the RMSF value of protein residue index Cα atoms of the complex structure. CID 10100411 (blue), CID 23266161 (red), CID118701104 (gray) and VP26 protein (blue) shows in separate colour.
3.10 Protein - ligand contact mapping
The interactions of protein with ligand is the fundamental factor to monitor which happened throughout the simulation during the drug design. It has been categorized by hydrogen bonds, hydrophobic, ionic and water bridge during the simulation (Fig. 7). In the study, Hydrogen bond plays the significant role in the periods of ligand binding, which stimulated the drug specificity, metabolization and adsorption. The backbone acceptor was occurred in 21 times whereas backbone donor was happened in eleven times during the protein and ligand interaction.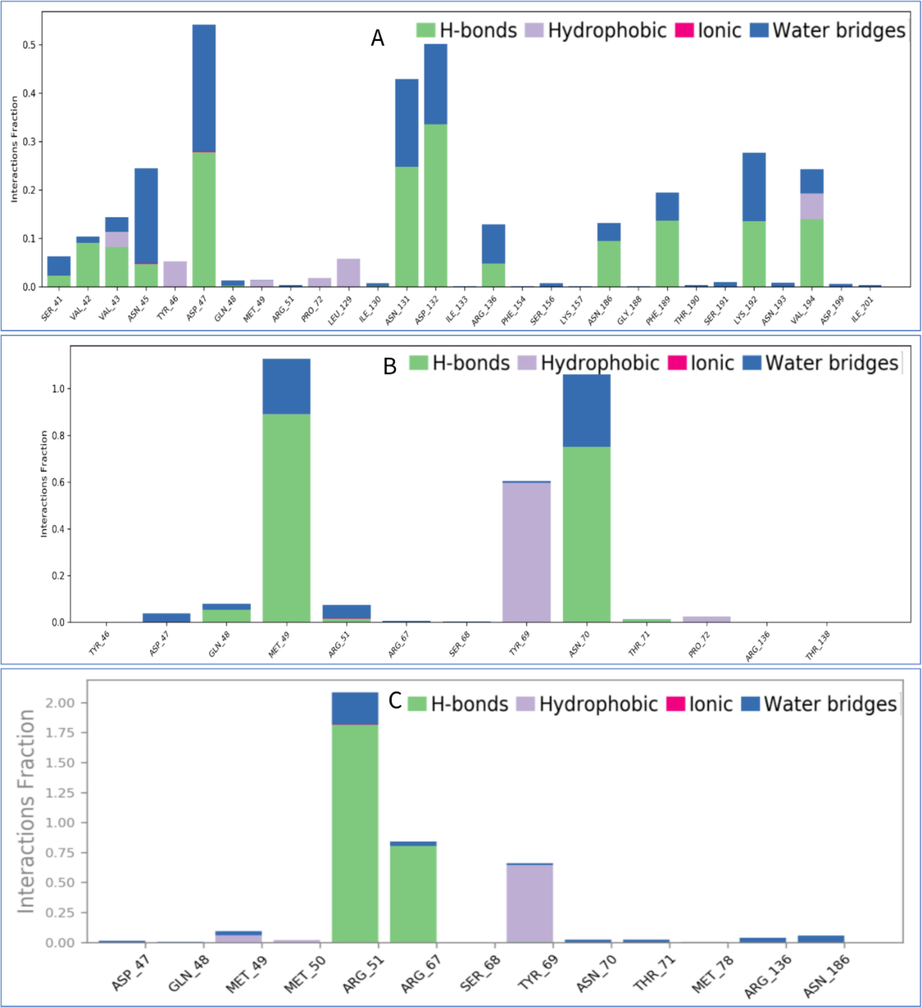
The bar charts indicate contact map of VP26 protein with the potential natural compounds from Withania somnifera, i.e., (A) CID10100411, (B) CID23266461, and (C) CID118701104 take out from 100 ns simulations.
4 Discussion
In the modern drug design, CADD has been performing as an indispensable tools for reducing cost, time duration and extra employment contribution during the modern drug discovery (Aljahdali and Molla, 2022). Therefore, it speeds up the research fields in broad spectrum through the efforts of biological and synthetic aspect. As a result, critical problems are solved within a short period in the globe such as Covid-19 vaccine invention thereby reducing the infection (Mostaghimi et al., 2021). Moreover, the CADD approach such as molecular docking, ADMET and dynamic simulation help to find out the best biological efficacy during the selection of drug like small molecules (Islam et al., 2022). CADD approach can not only document the specific diseases by the binding of ligand, and interacting and inhibiting the specific protein but also understand the behaviour mode between the ligand and specific target molecule. Dynamic simulation revealed the mechanism of complicated protein–ligand interaction, and molecular docking detected or predicted the binding mode of ligand and protein during the drug candidate selection process for a certain disease (JL and MK, 2018).
However, the 39 phytochemical compounds were selected and screened from the mangrove plant W. somnifera for targeting VP26 protein to inhibit WSSV infectious disease. Therefore, the highest binding affinity of compound has been chosen by the molecular docking approach after screening. In the present study, three compounds such as CID118701104 (A), CID10100411 (B), and CID23266461 (C) were selected from the 39 compounds on the basis of the higher binding score to the lower binding score −8.2, −8.1 and −7.9 kcal/mol, respectively. The protein and ligand interaction was determined by the binding of strong hydrogen and hydrophobic bond (Bulusu and Desiraju, 2020).
The metabolic kinetics of drug candidate as a small molecule inside of the body were focused by PK. ADME properties included the molecular weight and TPSA that can affected small molecules through the permeability of biological barrier (Daina et al., 2017). The molecular weight has gone down because of permeability, and TPSA has made the lower molecular weight more permeable. Moreover, lipophilicity can focus on the inorganic and aqueous phase of the selected molecules which influenced the absorption, transport, permeability, binding, and distribution in various organs and tissues along with endocrine system (Blokhina et al., 2016). The higher LogP values, resulting from the lower absorption were correlated with each other whereas the lower LogS value may affected the solubility of the selected molecules. The bilayer membranes have been closed when the hydrogen bond donors and acceptors were increased and decreased. The rotable bond should be within 10 that focused on the oral bioavailability of drug candidates. PK properties were documented from all selected compounds and further evaluation was performed during the study (Chung et al., 2015).
5 Conclusions
The infectious viral diseases, caused by white spot syndrome disease (WSSV) has been seriously devastating the shrimp farming industry since the last two decades through the mass killing of shrimps in the culture systems. It not only has wiped out the entire population within few days but also lead to huge economic losses in the world. However, there have not been developed any effective therapeutics against the viral infection till now. Therefore, this in silico technique has been advanced to find the natural and effective antiviral candidate against the hinder mature virion production and non-infectivity in the shrimp. Finally, it will not only provide new information about the matrix-like linker protein VP26 insight in the penaeid shrimp but also will open new avenue for the drug design of shrimp diseases in a significant and worthwhile approach.
Funding
This project was funded by the knowledge Excellence Program PhD, the Kingdom of Saudi Arabia Grant No (D1443-990-130).
CRediT authorship contribution statement
Mohammad Habibur Rahman Molla: Conceptualization, Methodology, Formal analysis, Writing – original draft. Mohammed Othman Aljahdali: Conceptualization, Supervision, Writing – review & editing, Funding acquisition.
Acknowledgments
The authors acknowledge with thanks to the Deanship of Scientific Research (DSR), King Abdulaziz University for technical support and also thanks to knowledge Excellence Program PhD, the Kingdom of Saudi Arabia for supporting and funding this project.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Aljahdali, M.O., Habibur, M., Molla, R., Filfilan, W.M., Nguyen, H., Zenger, K., 2021a. Whole genome sequence of the newly prescribed subspecies Oreochromis spilurus saudii: A valuable genetic resource for aquaculture in Saudi Arabia. J. Mar. Sci. Eng., 9, 506–9. https://doi.org/10.3390/JMSE9050506
- Immunoinformatics and computer-aided drug design as new approaches against emerging and re-emerging infectious diseases. Antivir. Drugs [Working Title] 2022
- [CrossRef] [Google Scholar]
- Population dynamics and fecundity estimates of Long-spined Black Sea Urchin Diadema savignyi (Audouin, 1890) from the Red Sea, Saudi Arabia. Saudi J. Biol. Sci.. 2022;29(9):103395.
- [CrossRef] [Google Scholar]
- Compounds identified from marine mangrove plant (Avicennia alba) as potential antiviral drug candidates against WDSV, an in-silico approach. Mar. Drugs. 2021;19:253.
- [CrossRef] [Google Scholar]
- Solubility, lipophilicity and membrane permeability of some fluoroquinolone antimicrobials. Eur. J. Pharm. Sci.. 2016;93:29-37.
- [CrossRef] [Google Scholar]
- Strong and weak hydrogen bonds in protein-ligand recognition. J. Indian Inst. Sci.. 2020;100:31-41.
- [CrossRef] [Google Scholar]
- First report of White spot syndrome virus in farmed and wild penaeid shrimp from Lagoa Dos Patos estuary, Southern Brazil. Braz. J. Microbiol.. 2011;42:1176.
- [CrossRef] [Google Scholar]
- Chung, T.D.Y., Terry, D.B., Smith, L.H., 2015. In vitro and in vivo assessment of ADME and PK properties during lead selection and lead optimization – Guidelines, benchmarks and rules of thumb. Assay Guid. Man.
- SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.. 2017;7:1-13.
- [CrossRef] [Google Scholar]
- Application of mathematical modeling and computational tools in the modern drug design and development process. Molecules. 2022;27:4169.
- [CrossRef] [Google Scholar]
- Islam, M.R., Awal, M.A., Khames, A., Abourehab, M.A.S., Samad, A., Hassan, W.M.I., Alam, R., Osman, O.I., Nur, S.M., Molla, M.H.R., Abdulrahman, A.O., Rajia, S., Ahammad, F., Hasan, M.N., Qadri, I., Kim, B., 2022. Computational identification of druggable bioactive compounds from Catharanthus roseus and Avicennia marina against colorectal cancer by targeting thymidylate synthase. Mol. 27, 2089. https://doi.org/10.3390/MOLECULES27072089.
- Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem.. 2018;26:2700-2707.
- [CrossRef] [Google Scholar]
- Karnila, R., Pratiwi, N., Sidauruk, S.W., 2020. The natural antioxidant potential of astaxanthin extracted from the whiteleg shrimp (Litopenaeus vannamei) carapace 13.
- Advances in the study of tegument protein VP26 in white spot syndrome virus. Aquac. Fish.. 2021;6:448-454.
- [CrossRef] [Google Scholar]
- Cathelicidins from the bullfrog Rana catesbeiana provides novel template for peptide antibiotic design. PLoS One. 2014;9(3):e93216.
- [CrossRef] [Google Scholar]
- Prevention of host-to-host transmission by SARS-CoV-2 vaccines. Lancet Infect. Dis. 2021
- [CrossRef] [Google Scholar]
- Mechanisms of disulfide bond formation in nascent polypeptides entering the secretory pathway. Cells. 2020;9(9):1994.
- [CrossRef] [Google Scholar]
- Computational assessment of MCM2 transcriptional expression and identification of the prognostic biomarker for human breast cancer. Heliyon. 2020;6(10):e05087.
- [CrossRef] [Google Scholar]
- Genetic variation in disease resistance against white spot syndrome virus (WSSV) in Liptopenaeus vannamei. Front. Genet. 2019:264.
- [CrossRef] [Google Scholar]
- Four major envelope proteins of white spot syndrome virus bind to form a complex. J. Virol.. 2009;83(9):4709-4712.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data (List of compounds identity, chemical name, two-dimensional (2D) structure of selected best three ligands, and control with their binding affinity were documented from the supplementary table (S1)) to this article can be found online at https://doi.org/10.1016/j.jksus.2022.102346.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1
List of compounds identity, chemical name, two-dimensional (2D) structure of selected best three ligands, and control with their binding affinity.
Supplementary data 2
Supplementary data 2




