

Translate this page into:
Formulation and evaluation of orodispersible tablet of taste masked doxylamine succinate using ion exchange resin
*Corresponding authors. Tel.: +91 9096482585 trusha_p10@rediffmail.com (T.Y. Puttewar), rupeshchikhale7@gmail.com (R.V. Chikhale)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Doxilamine orodispersible tablets were developed with considerable increase in drug release as compared to marketed formulations, seven formulations were developed and studied. The difference in drug release values was found to be 100.45 ± 1.89 and 56.47 ± 1.89, respectively. To prevent bitter taste and unacceptable odour of the drug, the drug was taste masked with weak cation exchange resins like Indion 234, Indion 204 and Indion 414. The drug was characterized according to different compendial methods, on the basis of identification by UV spectroscopy, pH, organoleptic properties and other tests. Among the three resins, one was selected for further studies i.e., Indion 234, because of high drug loading capacity. Drug–resin complex was prepared using batch method and effect of various processing parameters viz. drug–resin ratio, pH, temperature and drug concentration was studied to optimize the loading conditions. Maximum loading was obtained at drug–resin ratio 1:2, pH 5, temperature 50 °C and drug concentration 4 mg/ml. A successful taste masking of resinate was confirmed by time intensity method and also by taking drug release in 0.01 N hydrochloric acid and in simulated salivary fluid. The values of pre-compression parameters evaluated, were within prescribed limits and indicated good free flowing properties. The data obtained of post-compression parameters such as weight variation, hardness, friability, wetting time, water absorption ratio, content uniformity, disintegration time and dissolution and was found superior over conventional formulation. The F5 batch with disintegration time 25.24 ± 0.75 and dissolution 100.46% ± 3.78 was selected as optimized formulation. This was compared with conventional marketed formulation and was found superior. Batch F5 was also subjected to stability studies for three months and was tested for its disintegration time, drug contents and dissolution behaviour monthly. It was observed that the contents of the tablets remained the same. By an appropriate selection and combination of excipients it was possible to obtain orodispersible and taste masked tablets.
Keywords
Orodispersible tablets
Doxilamine
Taste masking
Ion exchange resin

1 Introduction
The term ‘Orodispersible Tablet’ as appears in European Pharmacopoeia (Suppl. 4.1, IV Ed.) is defined as “uncovered tablet for buccal cavity, where it disperses before ingestion”. They obviate the problem associated with conventional dosage forms, it has benefits like desired hardness, dosage uniformity, extremely easy administration and since no water is required for swallowing these tablets are suitable for geriatric, paediatric and travelling patients. These tablets display a fast and spontaneous de-aggregation in the mouth, soon after it comes in contact with saliva, dissolving the active ingredient and allowing absorption through all possible membrane it comes in contact during deglutition (Sharma et al., 2008).
In the recent past, several new advanced technologies have been introduced for the formulation of orodispersible tablets with very interesting features, like extremely low disintegration time, exceptional taste masking ability, pleasant mouth feel and sugar free tablets for diabetic patients (Shukla et al., 2009). The technologies utilized for fabrication of orodispersible tablets include lyophilization (Virely and Yarwood, 1990), moulding (Pebley et al., 1994), direct compression (Watanabe, 1995), cotton candy process (Myers et al., 1995), spray drying (Allen and Wang, 1996), sublimation (Koizumi et al., 1997), mass extrusion (Bhaskaran and Narmada, 2002), nanonization (Khan et al., 2007) and quick dissolve film formation (Khan et al., 2007). These techniques are based on the principles of increasing porosity and/or addition of superdisintegrants and water soluble excipients in the tablets (Biradar et al., 2006). The formulations prepared from these techniques differ from each other on the basis of the factors like mechanical strength of final product, drug and dosage form stability, mouth feel, taste, rate of dissolution of the formulation in saliva, rate of absorption from saliva and overall drug bioavailability and polymers (Kuchekar et al., 2003). Taste masking of the drug employing ion exchange resins (IER) has proved to be safe and effective method for formulation of various dosage forms.
In this paper we report the formulation and evaluation of orodispersible tablet of taste masked doxylamine succinate. Doxylamine succinate is an antihistaminic commonly used to prevent morning sickness in pregnant women. It is an extremely bitter drug therefore it is very essential to mask the bitter taste. It's formulation into simple tablet may induce vomiting, but formulation of taste masked doxylamine succinate in orodispersible tablet will give rapid action and will prevent morning sickness. Taste masking by ion exchange resin i.e., Indion 234 was employed because of its better drug loading and taste masking. Ion exchange resins have been increasingly used for the taste masking of bitter taste drug and help to prepare orodispersible tablets (Nandgude et al., 2007).
Ion exchange resins are solid and suitable in solubilised high molecular weight polyelectrolytes that can exchange their mobile ions of equal charge with the surrounding medium reversibly and stotiometrically. They are available in desired size ranges. Bitter cationic drugs can get adsorbed on to the weak cationic exchange resins of carboxylic acid functionally to form the complex which is not bitter. Further resinates can be formulated as lozenges, chewing gum, suspension or dispersible tablet and mask the taste (Rishi, 2004). Drug can be bound to the resin with the drug solution. Drugs are attached to the oppositely charged resin substrate or resonate through weak ionic bonding so that dissociation of the drug–resin complex does not occur under salivary pH conditions. This suitably masks the unpleasant taste and odour of drugs (Kaushik et al., 2004).
Taste is an important parameter in administering drugs orally. Undesirable taste is one of the important formulation problems that are encountered with many drugs. Administration of bitter drugs orally with acceptable level of palatability is a key issue for health providers. Proven methods for bitterness reduction and inhibition have resulted in improved palatability of oral pharmaceuticals (jeong et al., 2005). Taste masking is an essential requirement for fast dissolving tablets for commercial success. Taste masking of the active ingredient can be achieved by various techniques. Drugs with unacceptable bitter taste can be microencapsulated, such as Cefuroxime axetil can be microencapsulated in various types of acrylic polymers, Beclamide can be coacervated using gelatin. Macrolide antibiotics are having unacceptable taste (erythromycin and clarithromycin; Yajima et al., 1999) has reported successful taste masking using monoglycrides.
2 Materials and methods
2.1 Materials
All materials used in the present research were commercial samples. Active agent: Doxylamine succinate (Adroit Pharmaceuticals Pvt. Ltd., India), Pyridoxine HCL (Adroit Pharmaceuticals Pvt. Ltd., India); ION Exchange Resins: Indion 204, Indion 234 and Indion 414 (ION Exchange India Ltd., Mumbai, India); Excipients: VIVAPUR 102 (Microcrystalline Cellulose NF, Ph. Eur., JP), VIVASOL (Croscarmellose Sodium Ph. Eur., NF. JPE), VIVASTAR® P (Sodium Starch Glycolate, Ph. Eur.), Avicel PH 113 (Microcrystalline Cellulose USP/NF, EP,JP), Kollidon CL-SF (Super Fine Grade Crosspovidone USP/NF, EP, JPE), Lactopress® Anhydrous 250 (β-Lactose anhydrous USP/NF, EP, JP) and Ac–Di–Sol (Croscarmellose Sodium, NF, Ph, Eur., JPE) were gift by JRS PHARMA GmBH & CO. KG 73494, Rosenberg, Germany.
2.2 Preparation of tablets
The preparation of tablets was carried out after the analysis of drug samples, ion exchange resins, mixture formation and their analysis, drug loading studies, formulation and evaluation of Tablets (Rohm and Co, 2008).
2.2.1 Analysis of doxylamine succinate
The Doxylamine succinate was characterized according to different compendial methods and was found to be a white to creamy white crystalline powder with characteristic odour. Found to have a melting point in range of 103–106 °C and a pH of 5, λmax of 262 nm, all the findings matched the official reports.
2.2.1.1 Scanning of doxylamine succinate in 0.1 N hydrochloric acid
The solution containing 20 μg/ml of doxylamine succinate in 0.1 N hydrochloric acid was prepared and scanned over range of 200–400 nm against 0.1 N hydrochloric acid as a blank using Thermo Alfa Helios double beam UV spectrophotometer. The λmax was found to be 262.0 nm, which confirms to the reported value.
2.2.1.2 Preparation of dissolution medium for standard curves
In the present work, doxylamine succinate was estimated by UV spectrophotometry in distilled water, 0.01 N hydrochloric acid, 0.1 N hydrochloric acid and in simulated salivary fluid.
2.2.1.3 Preparation of standard calibration curve in distilled water
Various drug concentrations (5–50 μg/ml) in distilled water were prepared and the absorbance was measured at 262 nm.
For the standard curve, 100 mg of doxylamine succinate was accurately weighed and dissolved in 100 ml of distilled water, then 5 ml of the resulting solution was diluted to 100 ml with distilled water to make stock solution of concentration 50 μg/ml. Further serial dilutions were carried out with distilled water to get drug concentrations 5, 10, 15, 20, 25, 30, 35, 40, 45 and 50 μg/ml. The absorbance of dilutions was measured against distilled water as a blank at 262 nm using double beam UV/Visible spectrophotometer. The plot of absorbance Vs concentration was plotted and subjected to linear regression analysis. Drug was found to obey Beer Lambert's law in the concentration range of 5–50 μg/ml.
2.2.1.4 Preparation of standard calibration curve in simulated salivary fluid
Various drug concentrations (5–50 μg/ml) in simulated salivary fluid were prepared and the absorbance was measured at 262 nm (Clarke, 1986). For the standard curve, 100 mg doxylamine succinate was accurately weighed and dissolved in 100 ml of simulated salivary fluid, and then 5 ml of the resulting solution was diluted to 100 ml with simulated salivary fluid. Further serial dilutions were carried out with simulated salivary fluid to get drug concentrations 5, 10, 15, 20, 25, 30, 35, 40, 45 and 50 μg/ml. The absorbance of dilutions was measured against simulated salivary fluid as a blank at 262 nm using double beam UV/visible spectrophotometer. The plot of absorbance Vs concentration was plotted and subjected to linear regression analysis. Drug was found to obey Beer Lambert's law in the concentration range of 5–50 μg/ml.
2.2.2 Analysis of ion exchange resin (IER)
The IER is selected on the basis of drug nature and requirements of the formulation. The study was carried out among three different resins viz. Indion 204, Indion 234 and Indion 414 in order to screen most suitable resin for complexation with doxylamine succinate. Resinates were prepared using batch method. In each case, 100 mg doxylamine succinate in deionised water was stirred with 100 mg of resin using magnetic stirrer at 500 rpm. The amount of drug loaded at the end of 45 min was determined indirectly by estimating the amount remaining to be loaded in solution spectrophotometrically at 262 nm (United States Pharmacopoeia 28, 2005).
2.2.2.1 Effect of drug: resin ratio
The Indion 234 which showed highest amount of drug loading for ratio 1:1 was optimized for various drug:resin ratios. In each case, 100 mg of doxylamine succinate was stirred with varying amount of resin in deionised water using magnetic stirrer at 500 rpm. The amount of drug loaded at different time intervals was determined indirectly by estimating the amount remaining to be loaded in solution spectrophotometrically at 262 nm. A plot of mean of three determinations for percentage drug loaded as a function of time is plotted in Fig. 1. Results are displayed in Table 1. Results are the mean of three determinations.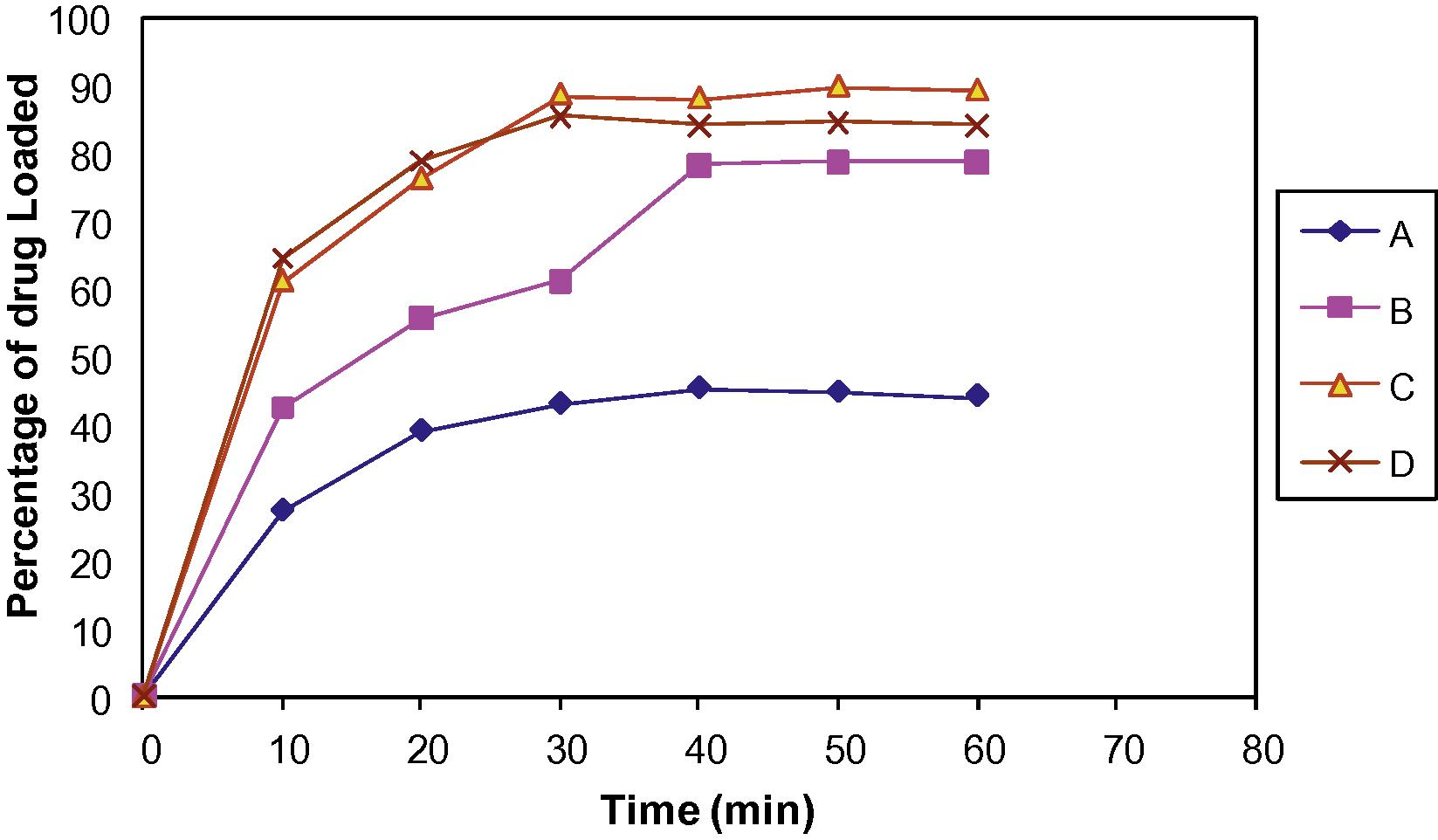
Effect of drug–resin ratio.
Time (min)
Percentage of drug loaded
A
B
C
D
00
00
00
00
00
10
27.46 ± 0.26
42.61 ± 0.46
61.17 ± 0.53
64.58 ± 0.26
20
39.2 ± 0.46
55.86 ± 0.70
76.7 ± 0.93
78.97 ± 0.46
30
43.17 ± 1.85
61.35 ± 1.39
88.63 ± 1.39
85.6 ± 0.71
40
45.45 ± 1.38
78.40 ± 2.32
88.06 ± 2.78
84.08 ± 4.64
50
44.88 ± 0.92
78.97 ± 2.78
89.77 ± 1.85
84.65 ± 0.92
60
44.31 ± 2.31
78.97 ± 3.24
89.2 ± 0.46
84.09 ± 5.10
2.2.2.2 Effect of pH on drug loading
The study was carried out at five pH values 3, 4, 5, 6 and 7. The pH was adjusted to desired value using 2 N citric acid. Solution of 100 mg doxylamine succinate drug was stirred with 200 mg of resin using magnetic stirrer at 500 rpm. The amount of drug loaded at different time intervals was determined indirectly by estimating the amount remaining to be loaded in solution spectrophotometrically at 262 nm. A plot of mean of three determinations for percentage drug loaded as a function of time at different pH was obtained.
2.2.2.3 Effect of temperature on drug loading
The study was carried out at four temperature conditions 30 °C, 40 °C, 50 °C and 60 °C. In each case, 100 mg of doxylamine succinate was stirred with 200 mg of resin in deionised water using magnetic stirrer at 500 rpm. The amount of drug loaded at different time intervals was determined indirectly by estimating the amount remaining to be loaded in solution spectrophotometrically at 262 nm. A plot of mean of three determinations for percentage drug loaded as a function of time was obtained.
2.2.2.4 Effect of drug concentration on drug loading
The study was carried out at three different concentrations 2 mg/ml, 4 mg/ml and 6 mg/ml. In each case, solution equivalent to 100 mg drug was stirred with 200 mg resin in deionised water using magnetic stirrer at 500 rpm. The amount of drug loaded at different time intervals was determined indirectly by estimating the amount remaining to be loaded in solution spectrophotometrically at 262 nm. A plot of mean of three determinations for percentage drug loaded as a function of time was obtained.
2.2.2.5 Optimized conditions
Following are the optimum conditions for the preparation of drug–resin complex of doxylamine succinate and are best suited for optimum loading
Drug:resin ratio: 1:2
pH: pH 5
Drug concentration: 4 mg/ml
Time: 30 min
2.2.2.6 Preparation of drug–resin complex
In batch process, 200 mg of activated resin was placed in a beaker containing deionised water and allow to swell for 30 min. Accurately weighed doxylamine succinate 100 mg was added and stirred for one hour. The mixtures were filtered and residue was washed with deionised water. DRC was then washed with sufficient quantity of deionised water for three times to remove loosely adsorbed drug from resinate surface. DRC was allowed to dry at room temperature and was stored in tightly closed container and used in further studies.
2.2.3 Studies on drug – complexes
2.2.3.1 Drug release from DRC
Drug release from DRC was determined using United States Pharmacopoeia (USP) type II dissolution apparatus. DRC equivalent to 30 mg of resinate was weighed accurately and added to 900 ml of 0.01 N hydrochloric acid and maintained at 37 °C. Drug release was performed at 50 rpm for 15 min. Aliquots of the medium were withdrawn at regular intervals, filtered and the absorbance determined on spectrophotometer. From absorbance values, percent drug dissolved at various time intervals was determined. A plot of mean of three determinations for percentage drug released as a function of time is plotted in Fig. 2 and results are displayed in Table 2. Results are mean of three determinations.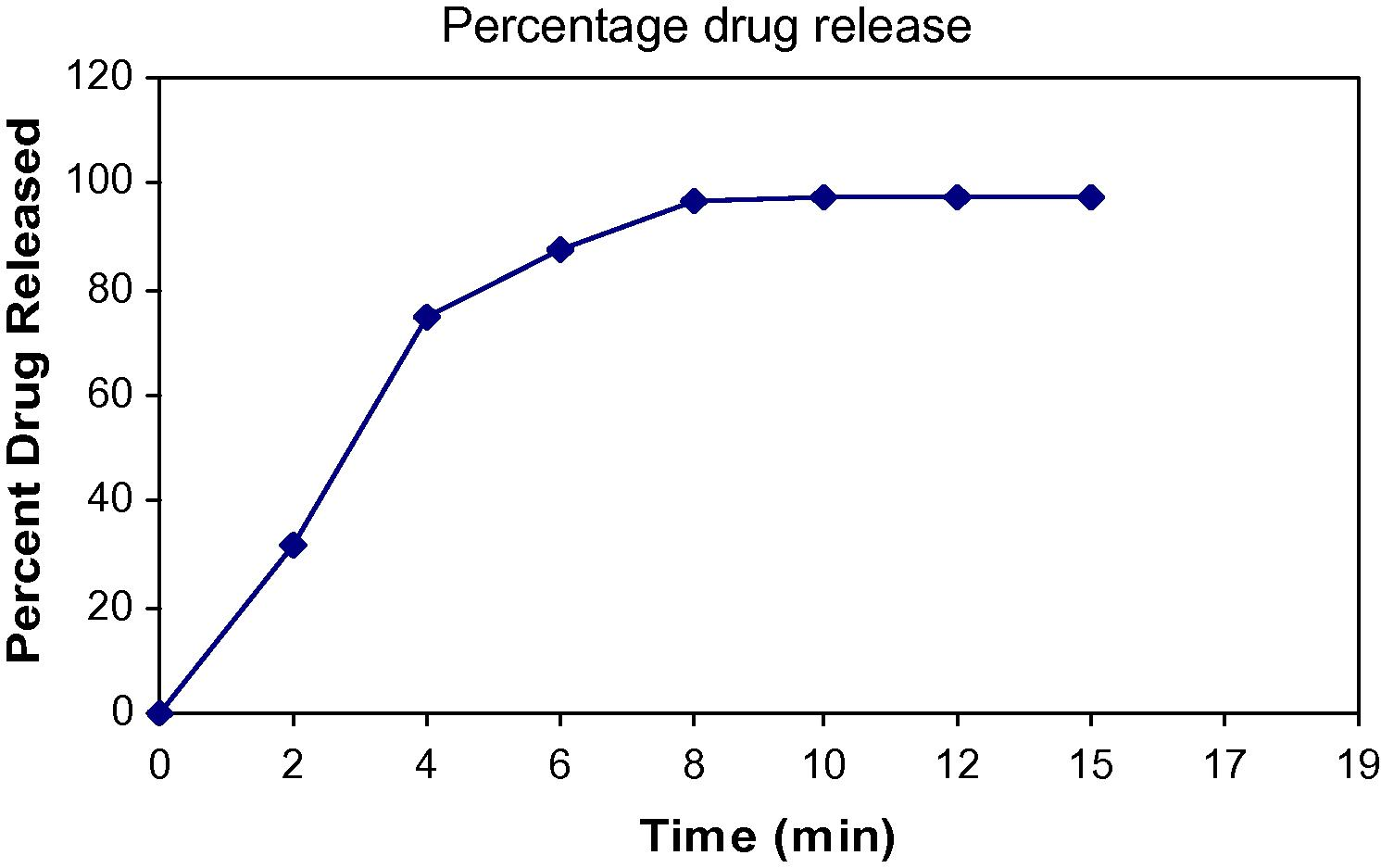
Drug release from DRC.
Sr. No.
Time (min)
Percentage drug release
1
0
0
2
2
31.77 ± 6.63
3
4
74.99 ± 7.55
4
6
87.33 ± 5.77
5
8
96.6 ± 3.93
6
10
97.37 ± 2.88
7
12
97.37 ± 2.18
8
15
97.37 ± 2.88
2.2.3.2 Taste panel evaluation of different batches of resinate
In this the different batches of resinate were tested to determine most suitable ratio of complex for successful taste masking.
The results (Table 3) showed excellent correlation with the evaluation of the taste panel employed. From above observations, Formulation C was selected for further study.
Batch
Taste-masked
Taste panel
A
Yes
Not acceptable worst than B
B
Yes
Not acceptable better than A
C
Yes
Maximum acceptability
D
Yes
Maximum acceptability
2.2.3.3 Taste panel evaluation for ‘Batch C’ resinate
Bitterness was quantitated by consensus of trained taste panel. A time intensity method was used, in which a sample equivalent to a normal dose was held in the mouth for 10 s (Fu et al., 1991). Bitterness level were recorded immediately, then spat out (Table 4). Results are mean of five determinations.
Sr. No.
Volunteers
10 s
1 min
2 min
5 min
10 min
15 min
1
1
0.7 ± 0.67
0.5 ± 0.31
0.2 ± 0.24
0.4 ± 0.58
0.2 ± 0.24
0
2
2
0.9 ± 0.58
0.3 ± 0.4
0
0.4 ± 0.58
0
0
3
3
0.6 ± 0.37
0.5 ± 0.44
0
0
0
0
4
4
0
0.3 ± 0.4
0
0
0
0
5
5
0
0.6 ± 0.37
0
0
0
0
6
6
0
0
0
0
0
0
7
7
0.4 ± 0.37
0
0
0.1 ± 0.2
0
0
8
8
0
0
0
0
0
0
9
9
0
0
0
0
0
0
10
10
0
0
0
0
0
0
A numerical scale was used with the following values: 0-tasteless, 0.5-very slight, 1-slight, 1.5-slight to moderate, 2-moderate, 2.5-moderate to strong, 3-strong, 3+-very strong.
2.2.3.4 ‘In vivo’ evaluation of drug complex
The said resinate was given to panel of healthy human volunteers for taste masking evaluation using time intensity method which shows satisfactory masking of taste (Table 5). Results are mean of five determinations.
Form of doxylamine succinate
Degree of bitterness
Time
10 s
1 min
2 min
5 min
10 min
15 min
Pure drug
2.2 ± 0.63
2.8 ± 0.24
2.5 ± 0.54
1.7 ± 0.97
1.1 ± 0.58
0.7 ± 0.50
Drug–resin complex (c)
1.1 ± 0.73
0.7 ± 0.67
0
0
0
0
2.2.3.5 ‘In vitro’ evaluation of drug content
Drug release from the DRC was also performed in 10 ml of pH 6.7 solution by adding drug complex equivalent to 10 mg of Doxylamine succinate to a test tube. The mixtures were filtered after shaking for 60 s. The filtrates were assayed for drug. Drug resinates are insoluble hence, even resinate of bitter drugs have virtually no taste. With the correct selection of ion exchange resin, the drug is not released in the mouth so that the patient does not taste the drug when it is swallowed. The percentage drug dissolved was found to be 1.96% ± 0.50 at a drug–resin complex ratio of 1:2.
2.2.3.6 X.R.P.D studies
The experiments were conducted on pure drug, pure resin, physical mixture of drug and resin and drug–resin complex for X-ray powder diffraction. The X-ray diffractograms were prepared by using Philips X’Pert PROP analytical XRD machine model PW1780.
The drug sample received from Adroit Pharmaceuticals complies with the compendial specifications for identification and other tests, and is suitable for present studies. Previously reported spectrophotometric methods of analysis of doxylamine succinate was found suitable for the present work and obeys Beer–Lambert's law in working range in all the media i.e., Distilled water, 0.01 N hydrochloric acid, 0.1 N hydrochloric acid and simulated salivary fluid.
The first step for purification of resin was done by washing with absolute ethanol, ethanol and water mixture and final washing with water to remove impurities associated with industrial scale, manufacturing or absorbed during storage or handling. For rapid release of drug, weak acid cation exchange carboxylic acid resins were tried. Preparation of resinate is normally done by two techniques (1) Bateh and (2) Column technique, but resinates were prepared using batch method.
Among three different resins viz. Indion 204, Indion 234 and Indion 414, the Indion 234 gave highest amount of drug loading (89.58% ± 0.26) and taste masking, because of its swelling and hydration properties hence Indion 234 was selected for further studies. Before designing a suitable delivery system, the main challenge is to optimize the conditions of resinate preparation, in order to obtain the desired drug loading in the resinates. Various processing parameters, viz. drug–resin ratio, pH, temperature and drug concentration, affected either rate or extent of loading of doxylamine succinate on Indion 234.
Study on effect of drug–resin ratio suggests higher drug loading at drug to resin ratio 1:1 and above. The plot of% drug loaded Vs time demonstrates that% uptake of drug from solution is highest for the ratio 1:2 i.e., 88.63% ± 1.39, for this reason drug–resin ratio 1:2 was selected as optimized ratio. Doxylamine succinate–Indion 234, complexation involves the exchange of ionizable drug and metal ions in resin, which in turn depends on the pKa of drug and resin. Such a mode of complexation between amino group of doxylamine succinate and –COO-K+ functionality of Indion 234 can be affected by the pH of the reacting media. The complexation was enhanced with increasing pH from 3 to 7. A maximum of 80.87% ± 0.26 wt/wt drug loading was obtained at pH 5 (i.e., at pKa of doxylamine succinate).
As shown in Fig. 3, as pH increased above 5 the percentage of drug loading decreased. The pH of the solution affects both solubility and the degree of ionization of drug and resin. The results can be attributed to the fact that doxylamine succinate has a pKa 4.4 and hence will have maximum solubility and complete ionization in this range. The decreased complexation at lower pH is due to excess H+ ions in the solution, which have more binding affinity to the –COO groups of resin and compete with the drug for binding.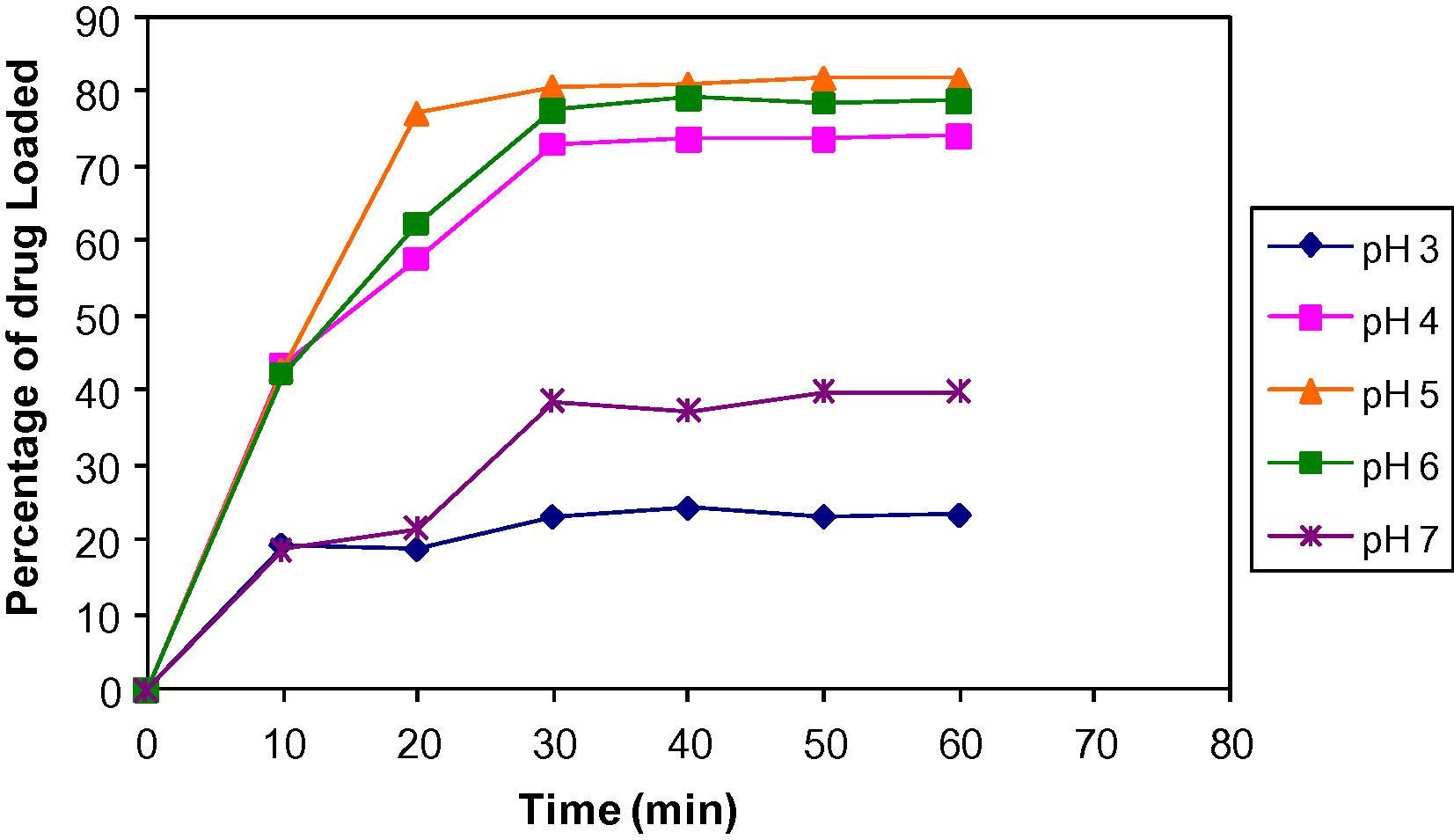
Effect of pH on drug loading.
The temperature also affected the complexation. Highest drug loading on resin occurred (90.71% ± 0.70) at temperature 50 °C. Increased temperature during complexation increases ionization of drug and resin. High temperatures may also cause swelling of resin, higher temperatures tend to increase the diffusion rate of ions by decreasing the thickness of exhaustive exchange zone. Drug concentration per ml of loading solution was found to affect the loading time and had no appreciable effect on extent of uptake. Drug loading was faster at higher drug concentration, probably because of efficient competition for binding site with other ions in solution. Thus higher drug concentration can be used to reduce the processing time and larger batches can be processed in relatively smaller vessels. The extent of drug loading was faster at drug concentration (4 mg/ml) i.e., 91.47% ± 0.46. Complexation between the drug and resin is essentially a process of diffusion of ions between the resin and surrounding drug solution.
The results reveal that a 30-min swelling time of Indion 234 in deionized water gave the maximum doxylamine succinate loading above 80%. The swelling and hydration properties of Indion 234 affect the rate of Ion exchange, which in turn affect the percentage drug loading. In unswollen resin matrix, the exchangeable groups are latent and coiled towards the backbone, hence less drug loading efficiency. The percentage of drug loading stirring time was studied. This study revealed that as time increased the KDM values increased rapidly up to 20 min, it is surprising to note the high exchange rate between 20 and 30 min. This finding may indicate the significant involvement of Vander Waals forces or chemisorption taking place along with exchange during complexation. Increasing the stirring time above 30 min did not further increase the KDM values. Hence 30 min contact time between drug and resin could be optimized to equilibrate the ion exchange process to achieve maximum drug loading. This study indicated that optimum ion exchange could be completed in a short period of 30 min.
Doxylamine succinate release from drug–resin adsorbate was observed using United States Pharmacopoeia USP 24 type II dissolution apparatus and drug release was 97.37% ± 2.88 within 30 min.
In vitro drug release in average salivary pH of 6.7 was less than 5% within 60 s. The presence of exchangeable ions of ionizable electrolytes in salivary fluid may be responsible for this release. Resinate is stable in salivary pH for a period of administration. The amount released is insufficient to impart bitter taste while the formulation passes through the mouth to further parts of gastrointestinal tract.
A drug–resin complex is made from the bitter drug and ion exchange resins. The nature of the complex is such that the average pH of 6.7 and cation concentration at about 40 meq/lit in saliva are not able to break the drug–resin complex but it is weak enough to be broken down by the hydrochloric acid present in the stomach, thus the drug–resin complex is absolutely tasteless and stable, with no aftertaste. The complexation of doxylamine succinate and Indion 234 produces amorphous tasteless drug resinate. Taste evaluation in volunteers found the DRC to be tasteless and agreeable with maximum acceptability.
The XRPD pattern of pure drug contained a number of sharp peaks, while the resin showed a diffused peak. The XRPD patterns of the physical dispersions showed simply the sum of the characteristic peaks were observed in the XRPD patterns for the complexes regardless of presence of drug. According to the data from XRPD, the molecular state of pure drug was crystalline, but that of the resin was amorphus. The molecular state of the drug prepared as drug–resin complexes was changed from the crystalline to the amorphous state. These results demonstrated that the entrapped molecule of drug was dispersed monomolecularly in the resin bead. In the case of physical dispersions of drug and Indion 234, drug molecules are outside the resin beads. Thus, DRC is amorphous in nature and shows faster dissolution due to improved solubility. FTIR spectra of pure drug, pure resin, physical mixture of drug and resin and resinate confirms the complexation between drug and resin, and support the findings of XRPD studies. The time intensity study adopted for taste evaluation reviled considerable masking of bitter taste of doxylamine succinate, which makes Indion 234 suitable for taste masking of doxylamine succinate.
2.2.4 Formulation of orodispersible tablets
Doxylamine succinate orodispersible tablets were prepared according to the formula given in Table 6. A total number of seven formulations were prepared. All the ingredients were passed through 60 mesh sieve separately and collected. The ingredients were weighed and mixed in a geometrical order. First MCC, Lactose and crospovidone were mixed together. Drug complex was then added and was mixed for 10–15 min. Finally to this blend aspartame and magnesium stearate were added and mixed further for 10–15 min (Fu et al., 1991). The tablets were then compressed using 10 mm size punches to get a tablet of 300 mg weight. The data for the selection of various superdisintegrants and their concentration for series S1–S12 are given as Supplementary data. Tablets are prepared in batch of 30; DRC: drug–resin complex.
Sr. No.
Table ingredients (mg)
Formulation
F1
F2
F3
F4
F5
F6
F7
1
DRC
30
30
30
30
30
30
30
2
Pyridoxine HCL
10
10
10
10
10
10
10
3
Microcrystalline cellulose (Vivapur 102)
244.32
–
122.16
162.88
183.24
81.44
61.08
4
Lactose (Lactopress Anhydrous 250)
–
244.32
122.16
81.44
61.08
162.88
183.24
5
Crospovidone (Kollidon CL-SF)
9.12
9.12
9.12
9.12
9.12
9.12
9.12
6
Magnesium stearate
2.80
2.80
2.80
2.80
2.80
2.80
2.80
7
Aspartame
3.75
3.75
3.75
3.75
3.75
3.75
3.75
8
Flavour (Vanilla)
q.s
q.s
q.s
q.s
q.s
q.s
q.s
Before tablet preparation, the mixture blend of all the formulations were subjected to pre-compression parameters like angle of repose, bulk density, tapped density,% compressibility and flowability. The orodispersible tablets prepared subjected to post-compression parameters like, content uniformity, hardness, friability, weight variation, dissolution and in vitro disintegration. Batches were prepared by direct compression method.
Direct compression is the preferred method for preparation of tablets. Current usage of the term “direct compression” is used to define the process by which tablets are compressed from the powder blends of active ingredient/s and suitable excipients. No pre treatment of the powder blends by wet or dry granulation is involved.
2.2.5 Evaluation of blend for orodispersible tablets
Blend was evaluated for flow properties.
2.2.5.1 Angle of repose
The flow characteristics are measured by angle of repose. Improper flow of powder is due to frictional forces between the particles. These frictional forces are quantified by angle of repose. Angle of repose is defined as the maximum angle possible between the surface of a pile of the powder and the horizontal plane.
By definition where h is the height of pile; r is radius of the base of the pile; θ is the angle of repose.
2.2.5.2 Bulk density
It is defined mathematically as
When particles are packed loosely, lots of gaps between particles are observed. Hence bulk volume increases making the powder light. Based on bulk volume, powders are classified as “light” and “heavy”. Light powders have high bulk volume. On the other hand, smaller particles may sift between the larger particles, the powder assume low bulk volume or high bulk density. Such powders are called heavy powders. The bulk density depends on particle size distribution, shape and cohesiveness of particles.
A powder (about 60 g) is passed through a standard sieve No. 20. A weighed amount (approximately 50 g) is introduced into a 100 ml graduated cylinder. The cylinder is fixed on the Bulk density. Apparatus and the timer knob is set (regulator) for 100 tappings. The volume occupied the powder is noted. Further, another 50 taps may be continued and the final volume is noted. For reproducible results, the process of tapings may be continued until concurrent volume is achieved. This final volume is the bulk volume. Then bulk density is calculated using equation. Bulk volume is also measured by dropping the cylinder (containing powder) onto a hard wooden surface 3 times from a height of 1 inch at 2 s intervals. Sometimes, to get an appropriate volume, the container has to be dropped or tapped 500 times.
2.2.5.3 Tapped density
Tapped density was determined by USP method II. The powder sample under test was screened through sieve No. 18 and 10 g of tablet bend was filled in 100 ml graduated cylinder of tap density tester (Electrolab, ETD 1020). The mechanical tapping of the cylinder was carried out using tapped density tester at a nominal rate of 250 drops per minute for 500 times initially and the initial tapped volume (Va) was noted. Tapping was proceeded further for additional 750 times and volume was noted. The difference between two tapping volumes was calculated. Tapping was continued for additional 1250 times if the difference is more than 2%. This was continued in increments of 1250 taps until difference between volumes of subsequent tapings was less than 2%. This volume was noted as, the final tapped volume (Vb).
The tapped density (Dt) was calculated in g/ml by the formula, where M is the weight of sample powder taken; Vb is tapped volume.
Determinations were carried out in 3 replicates. The mean value of three determinations are considered.
2.2.5.4 Compressibility
Carr's consolidation index: It is defined as:
This property is also known as compressibility. It is indirectly related to the relative flow rate, cohesiveness and particle size. It is simple, fast and popular method of predicting powder flow characteristics.
Fluff (poured) density is the ratio of mass of powder to the fluff volume. Fluff volume is the volume occupied by a certain mass, when gently poured into a measuring cylinder.
Tapped density is the ratio of mass of powder to the tapped volume. Tapped volume is the volume occupied by the same mass of the powder after a standard tapping of a measure.
Compressibility index can be a measure of the potential strength that a powder could build up in its arch in a hopper and also the ease with which such an arch could be broken.
Using a suitable adhesive, the base of a 10 ml tarred measuring cylinder is fixed to the standard rubber bung at the top of the 250 ml cylinder. A powder sample (about 5.0 g) is transferred into the tarred 10 ml cylinder with the help of a funnel. The 250 ml measuring cylinder is placed on the tapping apparatus. The initial volume occupied by the powder is denoted as V0.
The contents are tapped in the following order, 2, 4, 6, 8, 10, 20, 30 and 50 taps. After completing the tapings, the volume is denoted as V2, V4 … V50.
The powder is carefully collected from the cylinder and weighed (W)
2.2.5.5 Powder flowability
Before tablating the flowability of the mix ingredients of each formulation was studied. Although the pure doxylamine succinate was non flowable the addition of excipients resulted in a formation with percent compressibility between 12–16 and angle of repose between 20–30 indicating that flowability had significantly improved (Table 7). Results are mean of three determinations.
Sr. No.
Property
Formulations
F1
F2
F3
F4
F5
F6
F7
1
Angle of repose
25.56 ± 0.70
25.13 ± 0.78
26.25 ± 0.81
26.88 ± 1.77
26.61 ± 0.44
26.70 ± 1.63
26.45 ± 1.15
2
Bulk density gm/cm3
0.56 ± 0.011
0.56 ± 0.018
0.57 ± 0.008
0.57 ± 0.024
0.57 ± 0.016
0.57 ± 0.004
0.55 ± 0.020
3
Tapped density gm/cm3
0.65 ± 0.008
0.66 ± 0.024
0.67 ± 0.014
0.67 ± 0.008
0.67 ± 0.016
0.66 ± 0.012
0.65 ± 0.014
4
% compressibility
13.86 ± 1.42
13.7 ± 1.13
14.49 ± 0.63
13.96 ± 2.08
13.93 ± 0.77
13.54 ± 0.98
14.88 ± 1.98
5
Flowability
Good
Good
Good
Good
Good
Good
Good
2.2.6 Evaluation of tablets
The formulated orodispessible tablet were evaluated for different parameters like, weight variation (Borodkin and Sandburg, 1971), hardness, friability, wetting time (Kuchekar et al., 2004), water absorption ratio (Pisal et al., 2004), content uniformity and dissolution (Aly et al., 2005).
2.2.6.1 Weight variation
With a tablet designed to contain a specific amount of drug in a specific amount of tablet formula, the weight of the tablet being made is routinely measured to help ensure that a tablet contains the proper amount of drug. In practice, composite samples of tablets (usually 10) are taken and weighed throughout the compression process. The composite weight divided by 10, however, provides an average weight but contains the usual problems of averaged values. Within the composite sample that has an acceptable average weight, there could be tablets excessively overweight or underweight. To help alleviate this problem the United States Pharmacopoeia (USP)/National Formulary (NF) provides limits for the permissible variations in the weights of individual tablets expressed as a percentage of the average weight of the sample. The USP weight variation test is run by weighing 20 tablets individually, calculating the average weight, and comparing the individual tablet weights to the average. The tablets meet the USP test if no more than 2 tablets are out side the percentage limit and if no tablet differs by more than 2 times the percentage limit. The weight variation tolerances for uncoated tablets differ depending on average tablet weight.
2.2.6.2 Hardness
The hardness of a tablet is indicative of its tensile strength and is measured in terms of load/pressure required to crush it when placed on its edge. A number of handy hardness testers such as Mosanto type or Pfizer type are currently in use. Hardness of about 5 kg is considered to be minimum for uncoated tablets for mechanical stability. The hardness is a function of physical properties of granules like their hardness and deformation under load, binders and above all the compressional force. The hardness has influence on disintegration and dissolution times and is as such a factor that may affect bioavailabilities.
2.2.6.3 Friability
Generally it refers to loss in weight of tablets in the containers due to removal of fine particles from their surfaces. However, in wider sense chipping and fragmentations can also be included in friability. Friability generally reflects poor cohesion of tablet ingredients. Standard devices have been fabricated to measure friability. Generally such instruments, marketed as ‘Friability Test Apparatus’ or ‘Friabilators’, consist of a circular plastic chamber, divided into 2–3 compartments. The chamber rotates at a speed of 25 r.p.m. and drops the tablets by a distance of 15 cm. Preweighed tablets are placed in the apparatus, which is given 100 revolutions after which the tablets are weighed once again. The difference in the two weights represents friability. The weight loss should not be more than one per cent.
2.2.6.4 Wetting time and water absorption ratio
The procedures similar to those used Bi et al., where used to measure tablet wetting time and water absorption ratio. A piece of tissue paper folded twice was placed in a small petridish containing 6 ml of water. A tablet was kept on the paper and time required for complete wetting was measured. The wetted tablet was then weighed.
Water absorption ratio, R, was determined using the following equations: where, Wb = weight of tablet before water absorption, and Wa = weight of tablets after absorption.
2.2.6.5 Disintegration time
In vitro disintegration time of tables from each formulation were determined by using digital tablet disintegration apparatus. In vitro disintegration test was carried out at 37 ± 2 °C in 900 ml distilled water.
2.2.6.6 Content uniformity
Transfer 1 finely powdered tablet to a 100 ml volumetric flack containing 65 ml of 0.1 N hydrochloric acid, Shake frequently during a 10 min period, dilute with 0.1 N hydrochloric acid to volume, and mix. Allow the insoluble material to settle, and filter, discarding the first 20 ml of the filtrate. Dilute a Portion of the subsequent filtrate quantitatively and stepwise, if necessary, with 0.1 N hydrochloric acid to provide a solution containing approximately 25 μg of doxylamine succinate per ml. Concomitantly determine the absorbances of this solution and of a standard solution of USP doxylamine Succinate RS in the same medium having a known concentration of about 25 μg per ml in 1-cm cells at the wavelength of maximum absorbance at about 262 nm, with a suitable spectrophotometer, using 0.1 N hydrochloric acid as the blank. Calculate the quantity in mg, of C17H22N2O·C4H6O4 in the tablet taken by the formula:
in which; T is the labeled quantity in mg, of doxylamine succinate in the tablet, C is the concentration, in μg per ml of USP doxylamine succinate RS in the standard solution, D is concentration, in μg per ml doxylamine succinate in the solution from the tablet, based on the labeled quantity per tablet and the extent of dilution and AU and AS are the absorbances of the solution from the tablet and the standard solution, respectively, results are reported in Table 8. Results are mean of three determinations.
Sr. No.
Parameters
Formulation
F1
F2
F3
F4
F5
F6
F7
1
Weight variation
Passes
Passes
Passes
Passes
Passes
Passes
Passes
2
Hardness kg/sq cm
4.13 ± 0.24
4.23 ± 0.30
4.03 ± 0.26
4.16 ± 0.20
4.06 ± 0.28
4.0 ± 0.29
4.46 ± 0.12
3
Friability %
0.79 ± 0.012
0.76 ± 0.045
0.74 ± 0.036
0.77 ± 0.021
0.76 ± 0.044
0.72 ± 0.024
0.73 ± 0.059
4
Disintegration time (s)
32.52 ± 0.44
57.50 ± 1.04
46.55 ± 1.32
30.37 ± 0.66
25.24 ± 0.75
49.94 ± 1.34
57.09 ± 0.76
5
Content uniformity
97.92 ± 0.32
97.95 ± 0.07
98.28 ± 0.31
98.03 ± 0.05
98.89 ± 0.13
98.14 ± 0.22
98.32 ± 0.26
6
Wetting time (s)
10.04 ± 1.44
10.91 ± 0.90
11.95 ± 1.31
11.09 ± 0.61
10.60 ± 1.23
11.68 ± 0.40
13.34 ± 1.01
7
% Water absorption ratio
91.64 ± 0.32
95.64 ± 0.19
99.8 ± 0.13
103.75 ± 0.20
101.50 ± 0.32
72.8 ± 0.35
115.43 ± 0.60
2.2.6.7 Dissolution studies
The in vitro dissolution studies were carried out using USP apparatus type II at 50 rpm. The dissolution medium used was 0.01 N hydrocloric acid (900 ml) maintained at 37 ± 0.5 °C. Aliquots of dissolution media were withdrawn at different intervals and content of doxylamine succinate was measured by determining absorbance at 262 nm (Table 9). The dissolution experiments were conducted in triplicate. Not less than 80% (Q) of the labeled amount of C17H22N2O·C4H6O4 is dissolved in 30 min. The comparative results are shown as Fig. 4. Results are mean of three determinations.
Time (min)
Cumulative % drug release
F1
F2
F3
F4
F5
F6
F7
0
00
00
00
00
00
00
00
2
21.75 ± 11.33
29.47 ± 6.64
37.95 ± 13.23
41.81 ± 6.63
41.81 ± 4.75
40.27 ± 3.78
41.04 ± 11.39
4
52.62 ± 3.93
46.44 ± 12.30
51.84 ± 13.23
58.79 ± 5.67
67.27 ± 10.40
63.42 ± 7.56
57.25 ± 3.93
6
72.68 ± 9.44
68.05 ± 3.78
70.36 ± 11.33
74.99 ± 7.55
91.19 ± 5.67
81.16 ± 5.77
79.62 ± 3.77
8
83.48 ± 2.88
93.51 ± 1.89
95.05 ± 8.52
85.02 ± 2.88
98.14 ± 3.78
98.14 ± 1.89
93.51 ± 7.56
10
95.83 ± 3.78
93.51 ± 7.56
93.51 ± 3.78
98.14 ± 1.89
100.45 ± 1.89
98.91 ± 2.88
95.05 ± 2.88
12
95.82 ± 1.89
91.19 ± 5.67
93.51 ± 7.56
98.14 ± 3.78
101.23 ± 3.93
98.91 ± 1.09
95.82 ± 1.89
15
95.83 ± 7.56
93.51 ± 9.45
93.51 ± 5.67
98.91 ± 1.09
100.46 ± 3.78
99.68 ± 2.18
95.82 ± 5.67
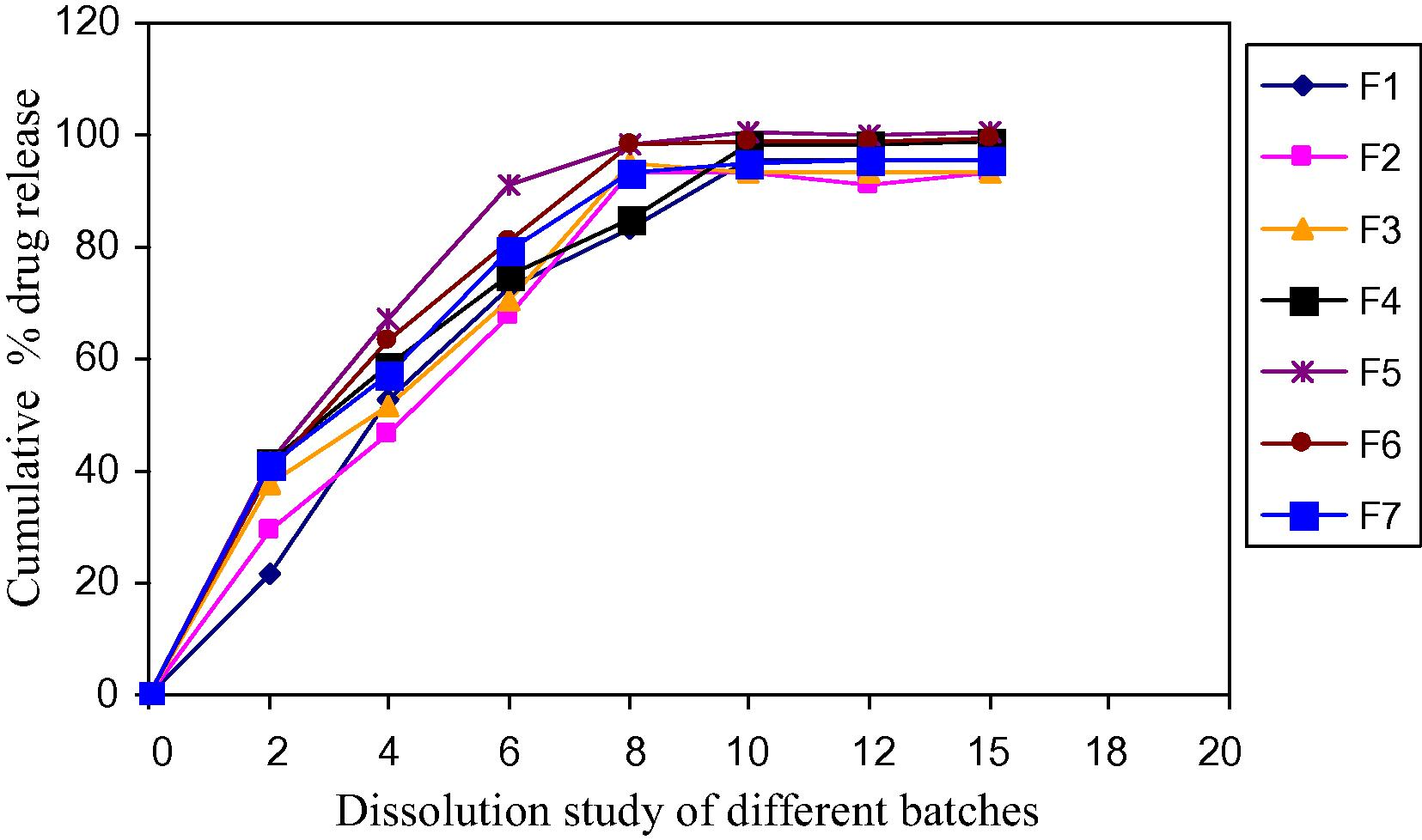
Dissolution study of different batches.
2.2.6.8 Comparison of optimized formulation with conventional marketed tablet
In vitro dissolution studies for batch F5 and conventional tablet were carried out using USP apparatus type II at 50 rpm, which shows that the drug release was more than 80% within 15 min which is better than conventional marketed tablet. The results are shown in Table 10, a plot of comparison is shown in Fig. 5.
Sr. No.
Time (min)
Cumulative % release from conventional marketed tablet
Cumulative % release from optimized batch
1
00
00
00
2
2
14.81 ± 10.00
42.59 ± 3.78
3
4
25.61 ± 6.64
69.59 ± 6.63
4
6
31.01 ± 5.00
90.42 ± 4.75
5
8
39.5 ± 8.93
95.82 ± 1.89
6
10
49.53 ± 7.56
99.68 ± 1.09
7
12
56.48 ± 3.78
99.68 ± 2.88
8
15
56.47 ± 1.89
100.45 ± 1.89
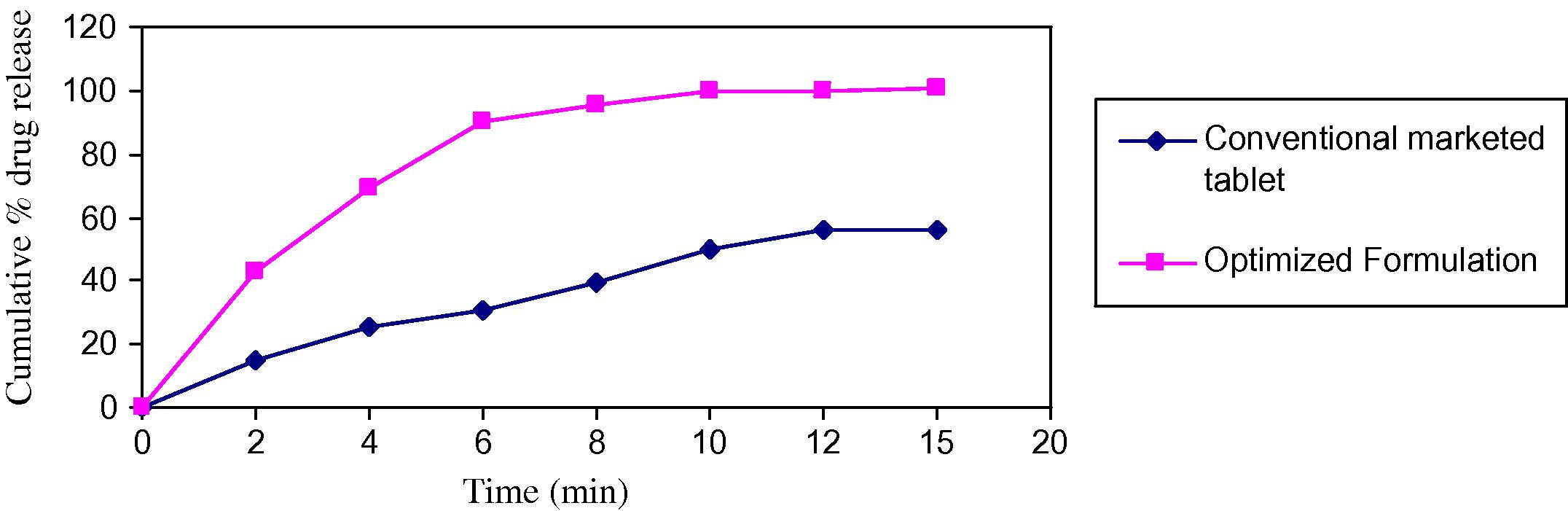
Comparison of optimised formulation with conventional marketed tablet.
2.2.6.9 Stability study
Stability study was conducted by storing tablets at 40 °C ± 2 °C/75 °C ± 5% relative humidity for three months (Gohel and Jogani, 2005). The content and dissolution behaviours from orodispersible tablets were tested monthly for three months (Tables 11–14).
Sr. No.
Evaluation
1 month
2 month
3 month
1
Hardness
3.96 ± 0.16
4.0 ± 0.21
4.1 ± 0.08
2
Disintegration time
24.73 ± 0.43
26.35 ± 0.77
28.36 ± 0.53
3
Content uniformity
98.17 ± 0.14
97.98 ± 0.08
99.96 ± 2.19
Sr. No.
Time (min)
Cumulative % drug release
1 month
2 month
3 month
1
0
0
0
0
2
2
18.66 ± 6.07
25.61 ± 3.93
23.29 ± 5.77
3
4
30.24 ± 9.70
40.27 ± 13.23
38.79 ± 6.64
4
6
57.25 ± 3.93
70.36 ± 5.67
54.93 ± 2.88
5
8
76.53 ± 2.88
87.34 ± 4.75
77.31 ± 5.66
6
10
94.28 ± 7.15
98.14 ± 3.78
92.74 ± 3.93
7
12
99.68 ± 2.88
101.22 ± 2.18
99.68 ± 1.09
8
15
99.68 ± 1.09
100.45 ± 1.89
99.68 ± 2.18
Sr. No.
Evaluation
1 month
2 month
3 month
1
Hardness
4.13 ± 0.12
4.06 ± 0.09
3.86 ± 0.04
2
Disintegration time
29.41 ± 1.16
27.75 ± 2.38
29.27 ± 2.73
3
Content uniformity
99.26 ± 2.19
99.07 ± 0.72
99.23 ± 0.98
Sr. No.
Time (min)
Cumulative % drug release
1 month
2 month
3 month
1
0
0
0
0
2
2
26.38 ± 1.89
37.95 ± 1.89
27.92 ± 12.15
3
4
43.36 ± 3.93
56.48 ± 3.78
58.79 ± 3.78
4
6
58.02 ± 6.64
76.54 ± 5.77
79.62 ± 3.77
5
8
81.16 ± 2.88
83.48 ± 1.08
98.91 ± 2.88
6
10
97.37 ± 2.88
98.14 ± 1.89
100.45 ± 3.27
7
12
98.14 ± 3.78
100.45 ± 1.89
100.46 ± 3.78
8
15
98.14 ± 5.67
100.46 ± 3.78
101.22 ± 2.18
Each tablets was individually weighed and wrapped in a aluminium foil and packed in black PVC bottle and put at above specified conditions in a heating humidity chamber for three months. After each month tablet sample was analysed for hardness, disintegration, time, dissolution and drug content. The results are shown below, comparative account is given in the form of plots (Figs. 6 and 7).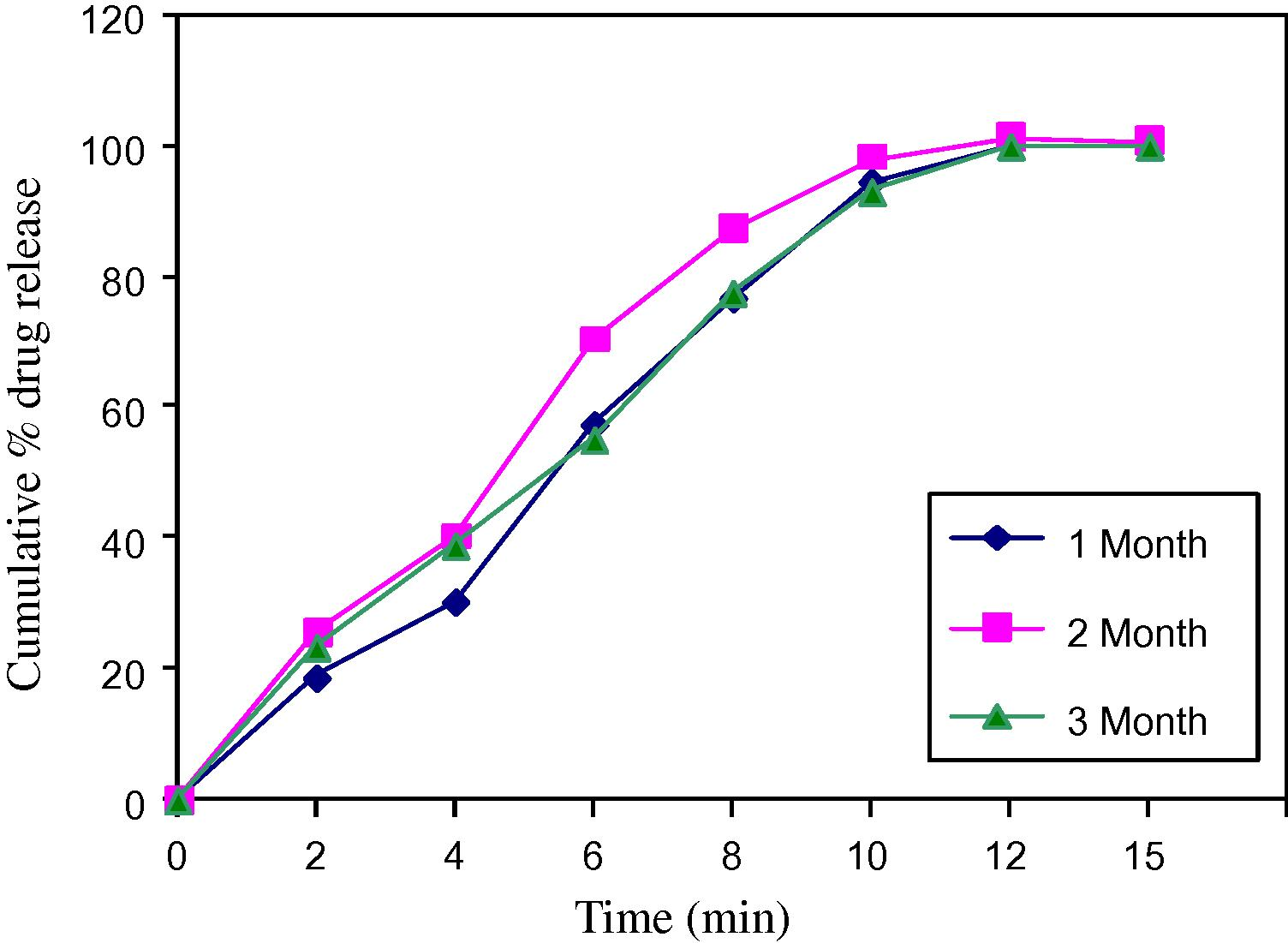
Dissolution profile for 40 °C.
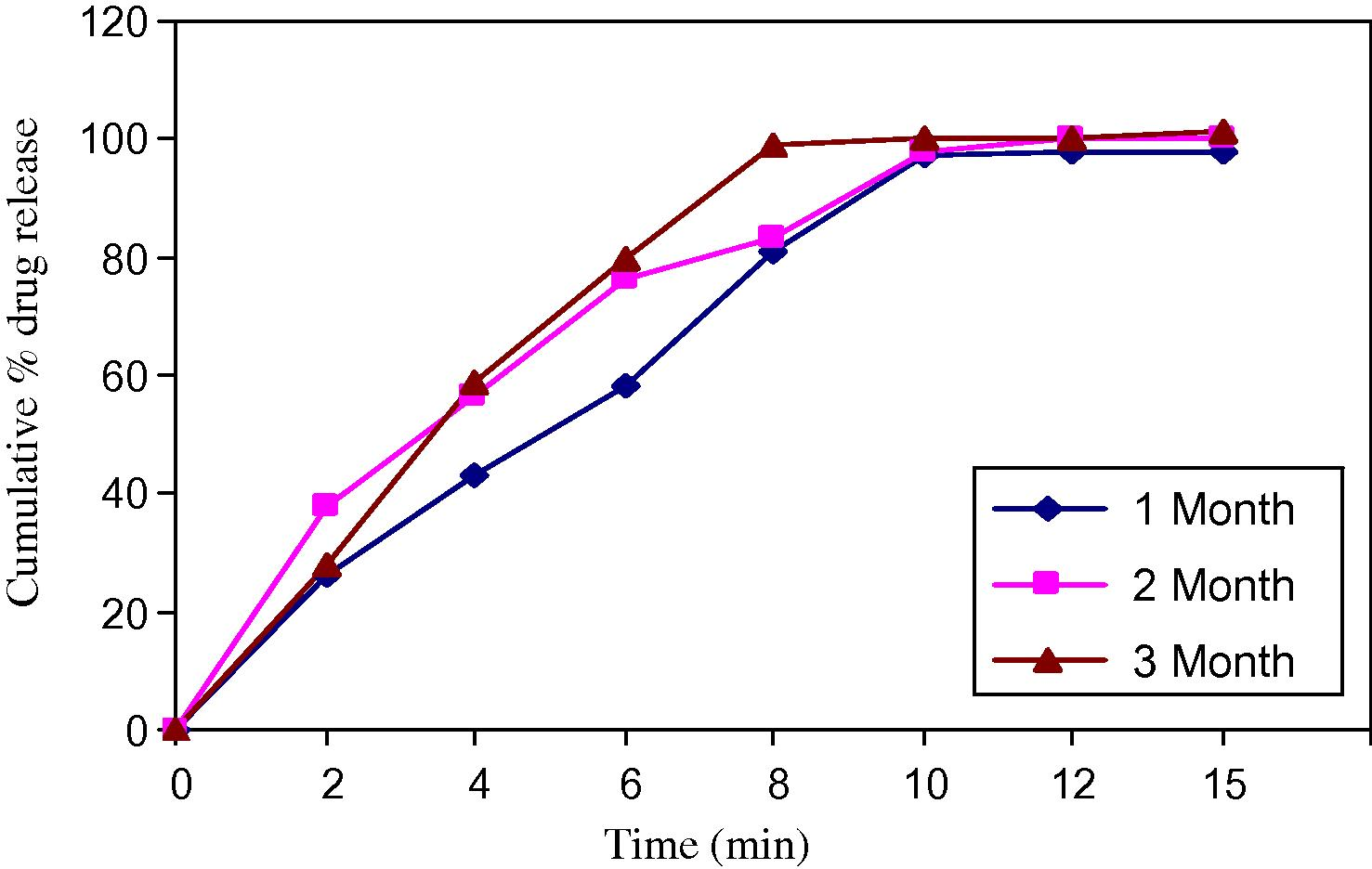
Dissolution profile for 75 °C.
3 Result and discussion
The formulation of orodispersible tablet was made by using doxylamine succinate–resin complex (resinate C). Batches S1–S7 were prepared by direct compression to select the disintegrant, from the results. It can be concluded that the tablets containing crospovidone (batch S6 and S7) exhibit quick disintegration time and followed by tablets containing croscarmelleose sodium and sodium starch glycolate. The probable reason for delayed the disintegration time of the tablet might be slow water uptake or more gelling tendency of croscarmellose sodium and sodium starch glycolate than crospovidone. Hence, crospovidone was selected as a disintegrant for the further studies. From the results it was obvious that the optimum concentration of crospovidone might be less than 10%. Batches S8–S12 were prepared to optimize the optimum concentration of crospovidone in order to obtain rapid disintegration at minimum concentration. Batches S6 (28.78 ± 0.48), S7 (39.30 ± 0.84) and S8 (26.55 ± 0.39) exhibited decrease in disintegration time and wetting time (10–12 s). But S8 had shown more decrease in disintegration time and wetting for this reason batch S8 was selected. In batch S7 disintegration time (39.30 ± 0.84) was found more than batch S8, such a behavior of superdisintegrants may be due to the blockade of capillary pores which prevents the entry of fluid into the tablet. For formulation of orodispersible tablet the blend was prepared and subjected to evaluation. The composition of blend of each batch is given in Table 6. Pyridoxine hydrochloride is added in the formulation because it helps in the healthy functioning of the nervous system and brain. Pyridoxine hydrochloride is involved in the production of hemoglobin in red blood cells and neurotransmitters like serotonin. It also improves energy production in the body and it is also essential for the normal reproductive process and healthy pregnancies.
The tablet blend of all the batches were evaluated for different derived properties viz.-angle of repose (between 25 and 27), Bulk density (between 0.55 and 0.57 gm/cm3), Tapped Density (0.65–0.67 gm/cm3), Compressibility index (between 13 and 15, and flowability (good). The results of Angle of repose and compressibility indicated that the flowability of blend is significantly good. Orodispersible tablets were prepared in batches F1–F7 and evaluated for tablet properties like, weight variation, hardness, friability, wetting time, water absorption ratio, content uniformity, disintegration time and dissolution.
All the tablets passed weight variation test as the percent weight variation was within the pharmacopoeial limits. Hardness were shown in the range of 4.0 ± 0.29–4.46 ± 0.12 kg/cm2 in all the formulations which indicated good mechanical strength with an ability to withstand physical and mechanical stress conditions while handling. In all the formulations, the friability value was less than 1% and meets the official limit. The results of disintegration of all the tablets were found to be within prescribed limits and satisfied the criteria of orodispersible tablet. The values were found to be in the range of 25.24 ± 0.75–57.50 ± 1.04. The percentage drug content of all the tablets was found to be between 97.92 ± 0.32 and 98.89 ± 0.13 of doxylamine succinate which was within acceptable limit. All the tablets prepared were subjected for release profile. The tablets prepared from crospovidone i.e., F1–F7 showed a drug release between 83.48 ± 2.88 and 101.23 ± 3.93. The wetting time and water absorption ratio was also in acceptable limit i.e., between 10.04 ± 1.44–13.34 ± 1.01 and 72.8 ± 0.35–115.43 ± 0.63. Among seven Batches, Batch F5 is selected as optimized batch because of its lowest disintegration time and highest drug release. In comparison, formulation F5 was compared with conventional marketed formulation. The drug release of marketed product and F5 formulation was found to be 56.47 ± 1.89 and 100.45 ± 1.89 at the end of 15 min. Stability was performed on formulation F5. Results for hardness, disintegration time, dissolution and content uniformity show no appreciable change up to 3 months of accelerated stability studies.
4 Conclusion
During the last decade, orodispersible tablets that make tablets disintegrate in the mouth without chewing and additional water intake has drawn a great deal of attention. Because of the increase in the average life span and the decline, with age, in swallowing ability, oral tablets administration to patients is a significant problem and has become the object of public attention. Solid dosage forms that can be dissolved or suspended with water in the mouth for easy swallowing are highly desirable for the pediatric and geriatric population, as well as other patients who prefer the convenience of readily administered dosage form. Organoleptic characteristics of pharmaceutical products, i.e., mainly appearance, odour and taste are essential factors in assessing the consumer acceptability, thereby the commercial success in the market. Thus taste masking of oral pharmaceuticals has become a potential tool to improve patient compliance and commercial success of the product. Ion exchange resins are solid and suitably insoluble high molecular weight polyelectrolytes that can exchange their mobile ions of equal charge with the surrounding medium. The resulting ion-exchange is reversible and stoichiometric with the displacement of one ionic species by another.
Molecular properties of resinate were studied using XRPD and FTIR, both suggested complexation between drug and resin. The change in crystalline form of drug to amorphous form due to monomolecular dispersion was suggested by these studies. The complexes were successfully formulated into orodispersible tablets. Three superdisintegrants were screened in order to determine most suitable superdisintegrant, among these, 9% w/w crospovidone was selected and tried for further studies. A total number of seven formulations were prepared by direct compression technique.
In the end of it can be concluded that pharmaceuticals complexed using ion exchange resins have shown improved organoleptic performance of pharmaceuticals and better patient compliance. This study shows an urgent need for a new dosage form which can improve patient compliance. For better taste masking effective techniques are being developed constantly in the pharmaceutical industry. Presently the use of these techniques depends on the nature of the drug, thus the use of cation exchange resin offers good taste masking of doxylamine succinate and its formulation into orodispersible table offers advantages over conventional tablet.
References
- Allen, L.V., Wang, B., 1996. Process for making a particulate support matrix for making a rapidly dissolving tablet. US Patent 5,587,180.
- Fast dissolving drug delivery system: a brief overview. Internet J. Pharmacol.. 2006;4(2):32-38.
- [Google Scholar]
- Polycarboxylic acid ion-exchange resin adsorbates for taste coverage in chewable tablets. J. Pharmaceut. Sci.. 1971;60(10):1523-1527.
- [Google Scholar]
- Clarke's, 1986. Isolation and Identification of Drugs in Pharmaceuticals, Body Fluids, and Post-mortem Material, second ed. The Pharmaceutical Press, London, p. 576.
- Fu, Lu Mou-ying et al., 1991. A polymer carrier system for taste masking of microlide antibiotics. Pharmaceutical Research 8(6), 706-711.
- A review of co-processed directly compressible excipients. J. Phram. Pharmaceut. Sci.. 2005;8(1):76-93.
- [Google Scholar]
- Frosta: a new technology for making fast-melting tablets. Expert. Opin. Drug Deliv.. 2005;2(6):1107-1116.
- [Google Scholar]
- Taste masking of ondensetron hydrochloride by polymer carrier system and formulation of rapid disintegrating tablets. AAPS PharmSciTech.. 2007;8(2):46-57.
- [Google Scholar]
- New method for preparing high porosity rapidly saliva soluble compressed tablets using mannitol with camphor, a subliming material. Int. J. Pharm.. 1997;152:127-131.
- [Google Scholar]
- Mouth dissolving tablets: a novel drug delivery system. Pharma. Times. 2003;35(June):7-9.
- [Google Scholar]
- Mouth dissolving tablet of salbutamal sulphate: a novel drug delivery system. Indian Drugs. 2004;41(10):592-597.
- [Google Scholar]
- Myers, G.L., Battist, G.E., Fuisz, R.C., 1995. Process and apparatus for making rapidly dissolving dosage units and product there from. PCT Patent WO 95/34293-A1.
- Mouth dissolving tablets: geriatrics and paediatrics friendly drug delivery system. Indian Drugs. 2007;44(6):471-473.
- [Google Scholar]
- Pebley, W.S., Jager, N.E., Thompson, S.J., 1994. Rapidly disintegrating tablet. US Patent 5,298,261.
- United State Pharmacopoeia 28/NF 23, (2005). Asian Edition, The Official compendia of Standards, United States Pharmacopoeial Convection Inc., Rockville, p. 702.
- Molecular properties of ciprofloxacin-indion 234 complexes. AAPS PharmSciTech.. 2004;5(4):262-275.
- [Google Scholar]
- Orally disintegrating tablets novel approach to drug delivery. Pharma. Rev.. 2004;34:36.
- [Google Scholar]
- Masking the taste of fast-disintegrating tablets. Drug Delivery Formulat.. 2008;4(5):109-111.
- [Google Scholar]
- Modified polysaccharides as fast disintegrating excipients for orodispersible tablets of roxithromycin. AAPS PharmSciTech.. 2008;9(1):1530-9932.
- [Google Scholar]
- Mouth dissolving tablets II: an overview of evaluation techniques. Sci. Pharm.. 2009;77:327-341.
- [Google Scholar]
- New compressed tablet rapidly disintegrating in the mouth using crystalline cellulose and a disintegrant. Biol. Pharm. Bull.. 1995;18:1308-1310.
- [Google Scholar]
- Yajima, T., Kuniashki, I., Shigeru, I., 1999. Taste Masking of Pharmaceutical Formulations. United States Patent 5,972,373.
Appendix A
Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jksus.2010.05.003.
Appendix A
Supplementary data
Supplementary data 1
Supplementary data 1
Supplementary tables and figures




