

Translate this page into:
Exploring the synergistic therapeutic potential of Morus alba extract in tuberculosis: A computational analysis
⁎Corresponding authors at: Department of Basic Dental and Medical Sciences, College of Dentistry, Ha’il University, Ha’il 2440, Saudi Arabia (S. Khan). sf.khan@uoh.edu.sa (Saif Khan)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Objectives
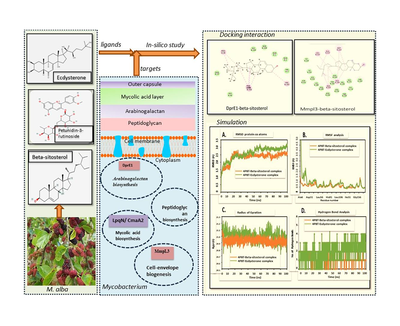
This study validates the synergistic impact hypothesis of Morus alba phytocompounds on Mycobacterium via molecular docking and simulation.
Methods
Phytocompounds (Petunidin-3-rutinoside, Beta-sitosterol, Ecdysterone, Quercetin-3′-glucoside, Quercitrin, Rutin, Scopolin) were tested against key metabolic proteins (MmpL3, DprE1, UgpABCE transporter, porins, OmpATb) of Mycobacterium. Additional proteins (CmaA2, oxidoreductase, FABH, Enoyl-ACP reductase, LpqN) were also included. Docked complexes were analyzed via MD trajectories (RMSD, RMSF, hydrogen bonds) over 100 ns.
Results
Binding affinity of phytocompounds with 1UUN (Beta-sitosterol: −9.12692, Ecdysterrone:-12.11162 kcal/mol) and 2KGS (Beta-sitosterol: −6.93) reflects the easy entry of phytocompounds. Phytocompounds interact within cells, inhibiting metabolic pathways and microbial growth. Beta-sitosterol (−7.1 kcal/mol) affects the mycolic acid transfer. Beta-sitosterol (−11.21 kcal/mol) and Ecdysterrone (−9.15 kcal/mol) affect the ribose oxidase pathway. Both compounds also show affinity with UgpABCE transporter. During simulation studies, results showed that the average protein RMSD for the 6MNA-Beta-sitosterol, 6MNA-Scopolin, 4P8T-Beta-sitosterol, and 4P8T-Ecdysterone complexes were 1.87 Å, 2.28 Å, 2.34 Å, and 2.61 Å, respectively. The stability of each complex in dynamic states is ensured by low and steady RMSD variance. In addition, we have endeavored to prove the hypothesis of synergistic application by incorporating our latest findings into the data.
Conclusions
Concept of synergistic impact of phytocompounds can be a promising source of treatment after proper lab validation.
Keywords
Natural compounds
Morus alba
Antitubercular activity
Docking
Interaction study

1 Introduction
Millions die annually from microbial infections, exacerbated by antibiotic ineffectiveness due to poor intracellular penetration and rising drug resistance (Tucker et al. 2021). Besides this, new resistant strains reduce medication efficacy or render them useless (Khameneh et al. 2019). Effective antibiotics demand continuous investment and innovative approaches like combination therapy, yet their success hinges on understanding synergistic interactions between drugs (Sharma et al., 2019). However, combining drugs may not boost efficacy without knowing complementary mechanisms or synergistic interactions. Comprehensive microbial and host biology knowledge is crucial for targeting essential microbial components and resistance mechanisms (Srivastava et al., 2021; Kumar et al., 2021).
Molecular understanding guides effective drug combinations that minimize resistance (Manna and Shil, 2020; Róg et al., 2021; Berríos-Caro et al., 2021). Computational methods like molecular docking and MD simulations streamline drug discovery, predicting interactions and optimizing treatments cost-effectively. Analyzing Morus alba's bioactive compounds computationally aims to uncover synergies against Mycobacterium tuberculosis (Mtb), offering promising candidates for experimental validation. This approach accelerates novel TB treatment strategies, emphasizing translational impact in pharmaceutical research.
2 Materials and methods
2.1 Hypothesis preparation
TB is primarily caused by Mtb, a Gram-positive, acid-fast bacterium known for its thick, lipid-rich cell envelope that contributes to drug resistance. Even when drugs penetrate this envelope, they can lose effectiveness due to enzymatic or structural modifications. Therefore, evaluating drug efficacy across multiple levels is crucial. This study focus on analyzing how selected metabolites interact with key proteins involved in various metabolic pathways of Mtb. Our objective was to assess the collective impact of these metabolites on Mtb's metabolism.
2.2 Selection of MTb proteins as target
The present study evaluates targets within the mycobacterial cell using five proteins sourced from the RCSB PDB database: Mtb porins (PDB ID: 1UUN), OmpATb (PDB ID: 2KGS) Mycolic acid transporter protein MmpL3 (PDB ID: 7NVH), Ribose oxidase DprE1 (PDB ID: 4P8T), and UgpABCE transporter (PDB ID: 4MFI)) (https://www.rcsb.org/).
2.3 Selection of ligands
From the previous investigation, 22 phytocompounds from Morus alba were screened against five target proteins of Mtb using in-silico tools. Out of these, seven phytocompounds exhibited promising activity against all five target proteins and were selected for further investigation.
2.4 Molecular docking studies
Molecular docking was conducted using Autodock, following the protocol described by Khan et al. (2023). Protein preparation included the removal of heteroatoms, addition of polar hydrogens, and Kollman charges, followed by saving in pdbqt format. Additionally, hydrogen-atoms were added to enhance ligand stability in the selected protein structures. Grid coordinates were defined to specify the docking grid-box. Docking experiments were performed under default settings in normal mode, and binding energy predictions (ΔG-score) were recorded for each ligand against the respective proteins.
2.5 Molecular dynamics (MD) simulation
MD simulation studies over 100 ns, were conducted for the best docking poses of beta-sitosterol, scopolin, and ecdysterone complexes with the proteins LpqN (PDB ID: 6MNA) and DprE1 (PDB ID: 4P8T) using the Schrödinger Desmond 6.9.137 MD simulation program (version 2022–1). The simulations were performed on a Z4 HP workstation with Ubuntu 22.04.2 LTS, Intel Xeon W-2245 @ 3.90 GHz, 8 cores, CUDA 12, and NVIDIA RTX A4000 GPU. MD simulations utilized the OPLS3e force field and SPC solvation model. Simulation details and experimental protocols followed previously published methodologies (Puri et al. 2023; Shaw, 2020-3). Desmond's Simulation Interaction Diagram (SID) was employed to analyze ligand binding orientation and stability based on MD trajectories (Desai et al. 2023).
3 Results and discussion
3.1 Background and hypothesis based on anti-mycobacterial activity of M. alba
The anti-inflammatory, liver and kidney-protective, hypotensive, diuretic, anti-cough, anticancer, and analgesic properties of Morus alba L. extract make it a compelling subject for study. Hence, Dried M. alba leaves were extracted with methanol, ethyl acetate, and chloroform (1:10 ratio) and tested for anti-tubercular activity using the Resazurin microtitre assay (REMA), as described in our previous study (Khan et al. 2023) and compared with the control drugs. Encouraging outcomes led to the hypothesis of synergistic effects of phytocompounds. The idea behind this approach is that diverse compounds can target multiple pathways in the pathogen, potentially preventing resistance development. We explored their potential for combination therapy, detailing our stepwise efforts below.
3.2 Target proteins
Porins and porin-like protein OmpATb were selected for study, as they facilitate hydrophilic nutrient passage across mycobacterial outer membranes. (Faller et al. 2004). The MmpL family facilitates lipid export from mycobacterial cytoplasm to periplasm (Melly and Purdy, 2019), another key focus of the study. The study also targets decaprenylphosphoryl-D-ribose oxidase and the UgpABCE transporter, enzymes crucial for mycobacterial arabinogalactan production (Suma et al. 2020). Additionally, five proteins from our previous study (Khan et al. 2023) were included to explore synergistic phytocompound effects: MTb Mycolic Acid Cyclopropane Synthase CmaA2 (PDB ID: 3HEM), oxidoreductase (PDB ID: 4OTK), A246F mutant of beta-ketoacyl-acyl carrier protein synthase III (FABH) (PDB ID: 2QO0), Enoyl-ACP(CoA) reductase (PDB ID: 2AQ1), and LpqN involved in cell envelope biogenesis (PDB ID: 6MNA).
3.3 Target ligand interaction studies via docking approach
In our earlier work, MTT assay was used to confirm the antitubercular activity of M. alba extracts before in-silico investigations to evaluate their activity against target proteins. Here, we extended our analysis to explore the synergistic effect hypothesis by comparing the activity of the top seven compounds from the prior study against five more proteins and summarized results in Table 1. Selected compounds already have some bioactivity like sitosterol and rutin for antimicrobial, anticancer, anti–inflammatory, hepatoprotective, lipid-lowering effect, anxiolytic & sedative effects, analgesic, immunomodulatory; Ecdysterone increases protein synthesis in skeletal muscle (Khan et al. 2023; Babu, & Jayaraman, 2020).
Ligand
Protein targets
Phytocompounds
study executed
docking energy (Kcal/mole)literature (Khan et al., 2023) docking energy (Kcal/mole)
S. No.
Name
ID
1UUN
2KGS
7NVH
4P8T
4MFI
3HEM
4OTK
2QO0
2AQI
6MNA
1.
Petunidin-3-rutinoside
101949843
−8.070
−1.65
−6.9
−7.81
−4.8
−8.2
−6.9
−9.0
−8.3
−7.8
2.
Beta-sitosterol
222284
−9.126
−6.93
−7.1
−11.21
−9.51
−9.0
−6.9
−7.4
−8.7
−6.7
3.
Ecdysterone
5459840
−12.111
−3.76
21
−9.15
−8.0
−7.5
−7.1
−8.2
−8.7
−6.3
4.
Quercetin-3′-glucoside
25203368
−8.250
−4.61
−6.6
−8.34
−6.86
−6.7
−7.6
−7.6
−7.6
−6.4
5.
Quercitrin
5280459
−8.308
−5.15
−6.9
−7.45
−6.92
−7.6
−7.6
−7.9
−7.4
−7.4
6.
Rutin
5280805
−9.832
−4.04
35.6
−7.05
−6.67
−7.8
−7.5
−9.1
−9.3
−6.9
7.
Scopolin
439,514
−8.068
−4.88
−2.7
−3.11
−6.63
−8.7
−6.9
−7.7
−7.4
−6.2
3.4 Entry of phytocompounds into Mycobacterium
The docking results of Mycobacterium proteins (1UUN, 2KGS, 7NVH, 4P8T, 4MFI) and the phytocompounds of Morus alba are summarized in Table 1. The Mtb cell's thick, hydrophobic wall poses initial penetration challenges. Reseaarch indicates that porins may aid hydrophilic antibiotics' diffusion through this barrier, facilitating cell entry (Gygli et al., 2017). Similarly, OmpATb in Mtb, changes pH, blocking hydrophilic drug entry thus inhibiting OmpATb with specific phytocompounds can facilitate the entry of other phytochemicals (Kwofie et al., 2020). Docking results of the selected phytocompounds range from −8.06 (Scopolin) to −12.11 (Ecdysterone) for porin and −6.93 (Beta-sitosterol) to −1.65 (Petunidin-3-rutinoside) for OmpATb, suggesting high affinity of these phytocompounds for these surface proteins. According to hypothesis, inhibition of OmpATb with Beta-sitosterol can facilitate the entry of other phytochemicals showcasing a synergistic effect. The interactions of ecdysterone, beta-sitosterol and rutin with porins are summarised in Fig. 1A and 1B suggesting the significant role of covalent interactions in the attachment of these phytocompounds to the porin protein.
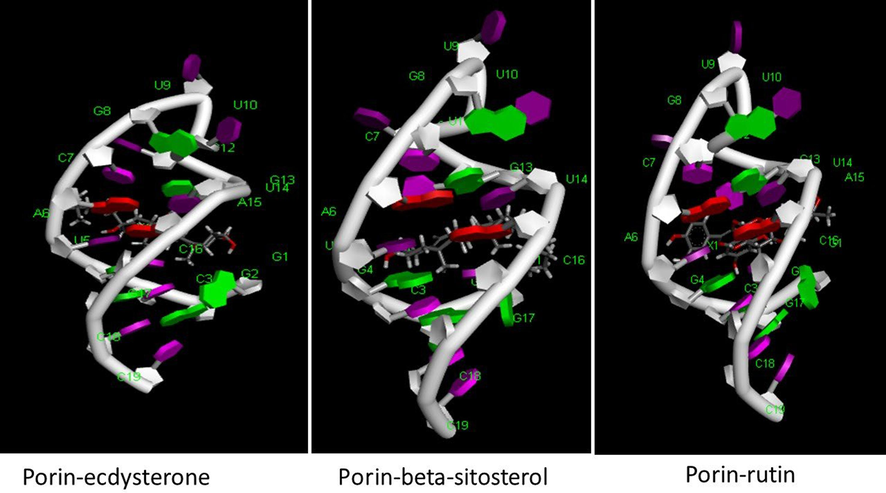
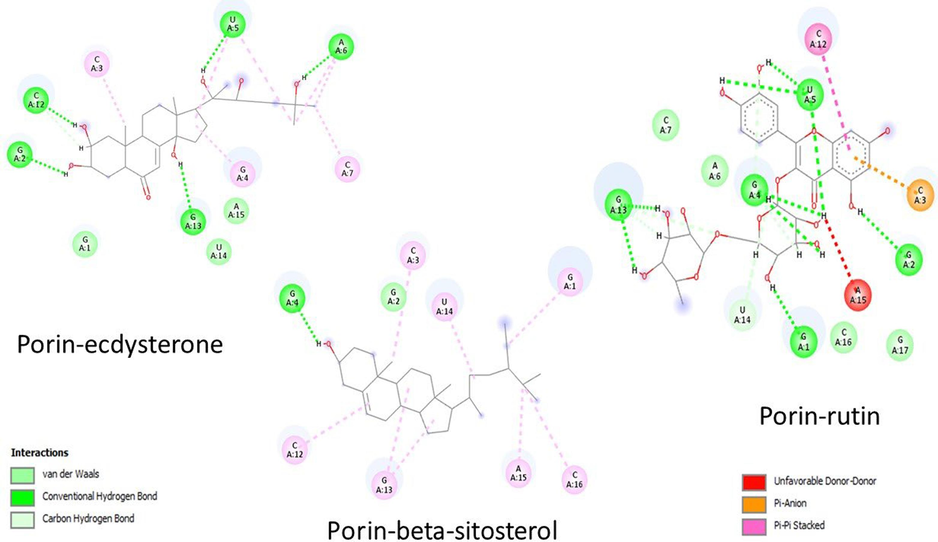
Hydrophobic compounds can pass through bacterial membranes by dissolving in the lipid bilayer, but hydrophilic compounds require protein channels like porins (MspA in M. smegmatis). Mtb uses porins like OmpATb in its outer membrane to facilitate hydrophilic solute entry (Niederweis, 2003). OmpATb is crucial for Mtb's survival in acidic environments. Kwofie et al. (2020) identified natural products that inhibit OmpATb, potentially disrupting Mtb's nutrient uptake and survival, offering new avenues for tuberculosis treatment development.
3.5 Action on different metabolic pathways
After entering, phytocompounds target vital biosynthetic pathways, crucial for the pathogen's structural and functional integrity. The absence of targeting proteins/pathways in the host or gut microbes will minimize the side effects (Maurya et al. 2022). The cell wall of Mtb, absent in humans and distinct from normal gut flora, is a prime target for drugs like isoniazid and ethionamide (Bolla, 2020).
3.6 Cell envelope biogenesis
The study also emphasizes Mycobacterial membrane protein large-3 (MmpL3; PDB ID: 7NVH), crucial for transporting precursors (trehalose monomycolates) in cell-envelope biogenesis. The docking results indicate that only Beta-sitosterol (−7.1) Petunidin-3-rutinoside (−6.9), Quercitrin (−6.9) and Quercetin-3′-glucoside (−6.6) exhibit a good affinity towards this protein, showcasing their potential to inhibit Mycobacterial cell envelope biogenesis. Fig. 2 summarizes the interaction of these phytocompounds with MmpL3, highlighting the significant role of covalent interactions. MmpL3 facilitates the transport of TMM across the inner membrane and has been a critical target for various preclinical agents (Bolla, 2020).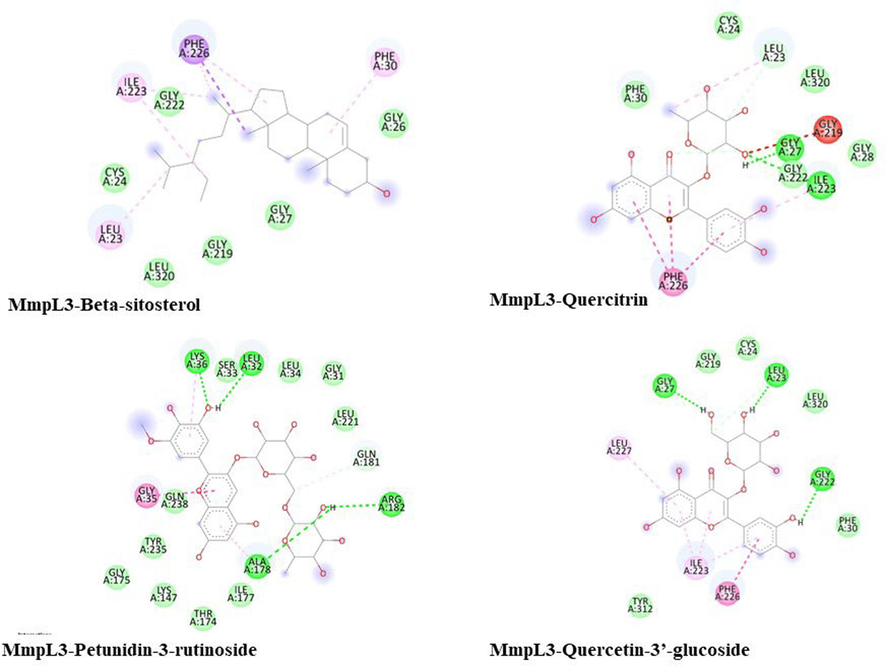
Interaction of beta-sitosterol, Petunidin-3-rutinoside, quercitrin and Quercetin-3′-glucoside with Mmpl3.
3.7 Arabinogalactan biosynthesis
DprE1 (Decaprenylphosphoryl-β-D-ribose 2′-epimerase), crucial for mycobacterial cell-wall synthesis, is a significant target. DprE1, a flavoprotein, catalyzes the oxidation of decaprenylphosphoryl-D-ribose (DPR) to decaprenylphosphoryl-2-ketoribose (DPX), which subsequently reduced to decaprenylphosphoryl arabinose (DPA) by enzyme, DprE2. Arabinosyltransferase converts DPA into arabinogalactan and lipoarabinomannan, key components of Mtb cell-wall (Mali et al., 2022). Out of seven, two phytocompounds- Beta-sitosterol and Ecdysterone with docking energy −11.21 and −9.15 Kcal/mole, respectively show their affinity to bind with DprE1 enzyme (4P8T) (Table 1) indicating their potential to inhibit the normal functioning of DprE1. In both cases, PHE and GLN residues of the active site form conventional hydrogen bonds (Fig. 3). Mali et al. (2022) reported that non-covalent inhibitors (2-carboxyquinoxalines) possess 2-carboxylate moiety crucial for hydrogen-bond formations with the sidechain of Lysine-418 and the hydroxyl group of Tyrosine-60. In the current work, tyrosine and leucine are also found to form hydrogen bonds with Petunidin-3-rutinoside, indicating a similar inhibition mechanism as for 2-carboxyquinoxalines.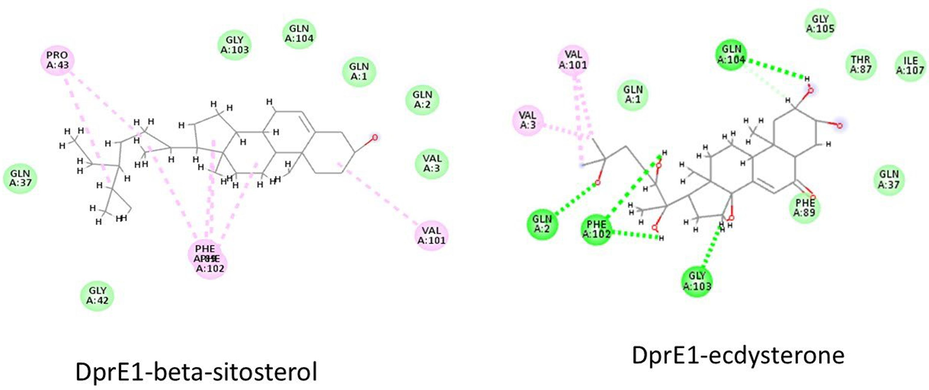
Interaction of beta-sitosterol and ecdysterone with DprE1.
3.8 Peptidoglycan biosynthesis
The absence of peptidoglycan biosynthesis in humans makes it a promising target to inhibit Mtb (Isa and Mohammed 2021). This study focuses on the final steps of the UDP-N-acetylmuramic acid formation, catalyzed by UDP-N-acetylenolpyruvoylglucosamine reductase (MurB). MurB is a Flavoprotein with a flavin adenine dinucleotide (FAD) binding structure, belonging to the superfamily of FAD-binding proteins (Murzin, 1996). Hence the peptidoglycan synthesis regulated by MurB is supported by the nutrient uptake facilitated by UgpABCE, highlighting a complex interplay between cell wall biosynthesis and nutrient acquisition in bacterial physiology (Soni et al., 2020). Ecdysterone with good binding affinity −9.51 kcal/mol with UgpABCE transporter (PDB ID: 4MFI) suggests indirect control on peptidoglycan biosynthesis.
3.9 Mycolic acid biosynthesis
Our previous research demonstrated the affinity of these compounds towards crucial proteins of Mycolic acid biosynthesis, including CmaA2 (3HEM: ΔG 6.7–9.0 kcal/mol), FaB (2QO0: ΔG 7.4–9.1 kcal/mol), and LpqN (6MNA: ΔG −6.2 to −7.8 kcal/mol). Petunidin-3-rutinoside, Beta-sitosterol, and Scopolin showed affinity for 3HEM, while Petunidin-3-rutinoside, Ecdysterone, and Rutin had affinity towards the 2QO0 protein. Additionally, these phytocompounds exhibited affinity towards 2AQI (ΔG −7.4 to −8.3 kcal/mol) enzyme having important role in FAS II cycle (Khan et al., 2023). β-sitosterol is well-documented for its antitubercular activity. Otari et al (2012) reported the antitubercular activity of ethanolic extract of plant Vitex negundo Linn containing betulinic acid, ursolic acid and β −sitosterol. Similarly, Sasikumar et al., (2018) reported the antimycobacterial potentials of quercetin and rutin.
3.10 MD trajectory analysis
The Glide docking protocol is flexible for small molecules but overlooks receptor structural dynamics in docking. Beta-sitosterol, Scopolin, and Ecdysterone were studied in complex with LpqN (6MNA), and DprE1 (4P8T) proteins to validate the docking results and assess their conformational stability. Each system underwent a 100 ns MD simulation, evaluating root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), and hydrogen bond interaction using MD trajectories. The values of each trajectory analyzing parameter including maximum, minimum, and average values are summarised in Table 2.
6MNA-Beta-sitosterol
6MNA-Scopolin
4P8T-Beta-sitosterol
4P8T-Ecdysterone
Root-mean-square deviation Å (RMSD)
Minimum
1.01
1.20
0.96
0.92
Maximum
2.48
3.00
2.80
3.24
Average
1.87
2.28
2.34
2.61
Root-mean-square fluctuation Å (RMSF)
Minimum
0.43
0.49
0.37
0.38
Maximum
3.89
5.69
4.77
6.33
Average
0.96
1.15
0.89
0.99
Radius of gyration Å (RGyr)
Minimum
20.82
20.96
20.73
20.81
Maximum
21.36
21.33
21.23
21.09
Average
21.04
21.19
21.00
20.93
Hydrogen Bonding
Minimum
1.00
1.00
1.00
1.00
Maximum
3.00
4.00
2.00
5.00
Average
0.66
1.19
1.13
1.96
The RMSD of alpha-carbon atoms helps to understand their stability (Kikiowo et al. 2022). Fig. 4A and 5A, for the 6MNA-Beta-sitosterol complex, the protein backbone's RMSD peaked at approximately 2.33 Å within the initial 24 ns, and stabilized around 2 Å for the remaining period. Similarly, Beta-sitosterol-4P8Tcomplex exhibited stable RMSD values around 3.2 Å throughout the simulation. For 6MNA-Scopolin Complex, an initial rise in RMSD around 2.97 Å occurred within 32 ns, stabilizing with minor fluctuations. The alpha-carbon of 4P8T-Ecdysterone complex exhibited stable variations with RMSD of below 3 Å with some initial fluctuations upto 21 ns.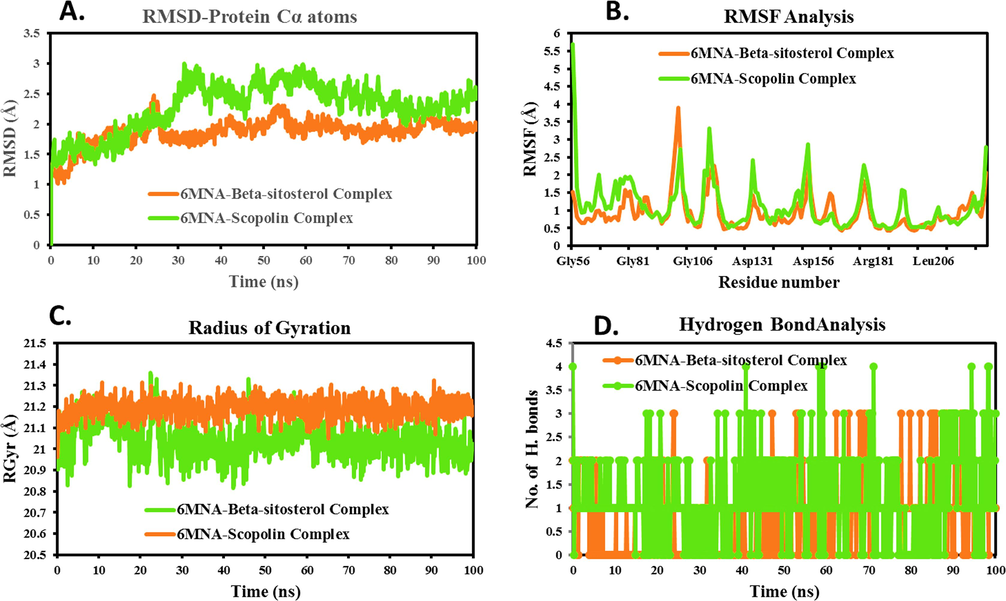
MD simulation Trajectory analysis of ligand-6MNA complexes A. Time dependent RMSD plot; B. Individual amino acids RMSF plot; C. Time dependent Hydrogen bond analysis; D. Time dependent Radius of gyration plot.
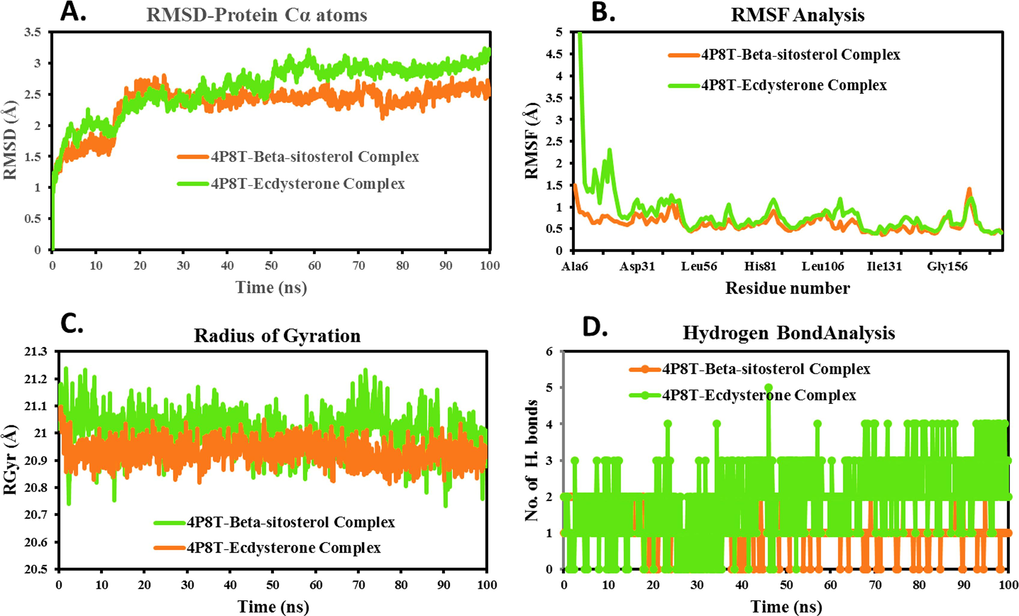
MD simulation Trajectory analysis of ligand-4P8T complexes A. Time dependent RMSD plot; B. Individual amino acids RMSF plot; C. Time dependent Hydrogen bond analysis; D. Time dependent Radius of gyration plot.
The initial RMSD rise may reflect structural changes or rearrangements in the protein backbone during phyto-compound interactions, possibly involving conformational adjustments or binding events. However, the subsequent RMSD stabilization indicates the complex settled into a relatively stable conformation, showing minor fluctuations in the simulation.
The 6MNA and 4P8T backbone complexes with phyto-compounds remained stable throughout the simulation, showing no significant deviations. The average protein backbone RMSD were 1.87 Å (6MNA-Beta-sitosterol), 2.28 Å (6MNA-Scopolin), 2.34 Å (4P8T-Beta-sitosterol), and 2.61 Å (4P8T-Ecdysterone). The maximum RMSD across all frames was 3.24 Å, indicating no extreme deviation during the MD simulation. The low average and RMSD support the stability of each complex in dynamic states (Fig. 4A and 5A).
Pandey et al. (2022) elucidated the role of amino acids in maintaining protein–ligand complexes during dynamic phases. The amino acid deviation of individual 6MNA and 4P8T proteins are shown in Fig. 4B and 5B. In the 6MNA protein complex, a maximum fluctuation of 3.89 Å and 3.321 Å is detected in Ser100 and Ala113, respectively. The visual analysis of MD simulation trajectories suggests that both Beta-sitosterol and Scopolin engaged in significant binding interactions with residues, Ala184, Ala203, Ala217, Ile121, Ile220, Leu201, Leu213, Met214, Ser154, Thr182, and Thr210 of the 6MNA protein. Residues with low RMSF values (0.43 Å and 1.95 Å) indicate stable Beta-sitosterol and Scopolin structures during simulation, suggesting higher protein stability. Amino acids of the 4P8T protein, including, Ala244, Asn385, Gln334, Gln336, Gly117, His132, Leu115, Leu317, Lys134, Lys418, Phe320, Pro116, Pro316, Ser228, Trp230, Trp296, Tyr297, Tyr303, Tyr314, and Val365, interact with Beta-sitosterol and Ecdysterone which have RMSF values less than 2.5 Å.
From Table 2, the average RMSF of 6MNA-Beta-sitosterol, 6MNA-Scopolin, 4P8T-Beta-sitosterol, and 4P8T-Ecdysterone complexes was found to be 0.96, 1.15, 0.89, 0.99 Å respectively. Loop regions distant from the binding pocket show higher fluctuations in RMSF. These residues lack binding interactions with the studied phyto-compounds, as indicated by their above-average values, explanation, and Fig. 4B and 5B. This suggests that during dynamic phases, all amino acids in the 6MNA and 4P8T proteins consistently and strongly bind the phyto-compounds.“.
Radius of gyration (RGyr) assesses structural compactness in complexes by measuring mass concentration distance from an axis, crucial for understanding macromolecules' 3D structure, folding behavior, and conformational changes during simulations. (Maurya et al. 2022; Patel et al. 2023). Large RGyr values correspond to more flexible conformations, while small RGyr values indicate more constrained conformations (Ayipo et al. 2022). The minimal Deviations for 6MNA (20.82–21.36 Å) and 4P8T (20.73–21.23 Å) complexes with phyto-molecules (Fig. 4C, 5C) suggest stable structures with limited conformational changes during simulations, indicating protein stability. RGyr deviation for 6MNA-Beta-sitosterol, 6MNA-Scopolin, 4P8T-Beta-sitosterol, and 4P8T-Ecdysterone, was 0.36, 0.54, 0.51, and 0.29 Å, respectively suggesting the firmly defined structure of proteins with few conformational changes.
In MD simulations, hydrogen bonds (HBs) ≤ 3 Å between ligands and proteins enhance complex stability (Tabti et al. 2023). Beta-sitosterol and Scopolin showed significant HB formation profiles over 100 ns, influencing binding affinity by establishing multiple specific interactions, as depicted in Fig. 4D and 5D.
4 Conclusion
In-silico investigation identified Morus alba phytocompounds (e.g., Petunidin-3-rutinoside, Beta-sitosterol) with strong binding affinity to Mtb targets: MmpL3 (7NVH), DprE1 (4P8T), UgpABCE transporter (4MFI), and OmpATb (2KGS). They also bind CmaA2 (3HEM), oxidoreductase (4OTK), FABH (2QO0), ACP(CoA) reductase (2AQ1), and LpqN (6MNA). This suggests their potential for combined therapy against Mtb, pending lab validation, highlighting the benefits of targeting multiple pathways for improved treatment outcomes.
Funding
This research has been funded by Scientific Research Deanship at university of Ha’il − Saudi Arabia through project number MDR-22020.
CRediT authorship contribution statement
Mahvish Khan: Writing – original draft. Saif Khan: Funding acquisition, Resources. Freah L Alshammary: Software. Urvashi Goyal: Writing – original draft, Methodology. Vineeta Singh: Supervision, Writing – review & editing, Conceptualization. Iqrar Ahmad: Validation. Harun Patel: Validation. V.K. Gupta: Writing – review & editing. Shafiul Haque: Writing – review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Computational modelling of potential Zn-sensitive non-β-lactam inhibitors of imipenemase-1 (IMP-1) J. Biomol. Struct. Dyn. 2022:1-21. Advance online publication.
- [CrossRef] [Google Scholar]
- An update on β-sitosterol: A potential herbal nutraceutical for diabetic management. Biomed. Pharmacother.. 2020;131:110702
- [Google Scholar]
- Competition delays multi-drug resistance evolution during combination therapy. J. Theoret. Biol.. 2021;509:110524
- [Google Scholar]
- Targeting MmpL3 for anti-tuberculosis drug development. Biochem. Soc. Trans.. 2020;48(4):1463-1472.
- [Google Scholar]
- The structure of a mycobacterial outer-membrane channel. Sci.. 2004;303(5661):1189-1192.
- [Google Scholar]
- Antimicrobial resistance in Mycobacterium tuberculosis: mechanistic and evolutionary perspectives. FEMS Microbiol. Rev.. 2017;41(3):354-373.
- [Google Scholar]
- Molecular docking and dynamic simulation of UDP-N-acetylenolpyruvoylglucosamine reductase (MurB) obtained from Mycobacterium tuberculosis using in silico approach. Netw. Model. Anal. Health Inform. Bioinform.. 2021;10(1):40.
- [Google Scholar]
- Review on plant antimicrobials: a mechanistic viewpoint. Antimicrob. Resis. Infec. Cont.. 2019;8(1):1-28.
- [Google Scholar]
- In silico analysis to identify potential antitubercular molecules in Morus alba through virtual screening and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2023:1-8.
- [Google Scholar]
- Molecular dynamics simulation and pharmacokinetics studies of ombuin and quercetin against human pancreatic α-amylase. J. Biomol. Struct. Dyn. 2022:1-8. Advance online publication. Doi: 10.1080/07391102.2022.2155699
- [Google Scholar]
- In silico studies reveal antiviral effects of traditional Indian spices on COVID-19. Curr. Pharm. Des.. 2021;27(32):3462-3475.
- [Google Scholar]
- Molecular docking and dynamics simulations studies of OmpATb identifies four potential novel natural product-derived anti-Mycobacterium tuberculosis compounds. Comput. Biol. Med.. 2020;122:103811
- [Google Scholar]
- Identification of hydantoin based Decaprenylphosphoryl-β-d-Ribose Oxidase (DprE1) inhibitors as antimycobacterial agents using computational tools. Sci. Rep.. 2022;12(1):16368.
- [Google Scholar]
- A review on potential drug delivery system as a treatment of intercellular bacterial infection. J. Pharmacovig. Drug Res.. 2020;1(2):13-23.
- [Google Scholar]
- In-silico study reveals potential antitubercular drug targets unique to Mycobacterium tuberculosis H37Rv. Minerva Biotechnol. Biomol.. 2022;34(2)
- [Google Scholar]
- Melly, G., and Purdy G. E., 2019. “MmpL Proteins in Physiology and Pathogenesis of M. tuberculosis.” Microorganisms 7.3 (2019): 70.
- Structural classification of proteins: new superfamilies. Curr. Opin. Struct. Biol.. 1996;6(3):386-394.
- [Google Scholar]
- Mycobacterial porins–new channel proteins in unique outer membranes. Mol. Microbiol.. 2003;49(5):1167-1177.
- [Google Scholar]
- Effect of hydroalcoholic extract of Vitex negundo Linn. leaves on learning and memory in normal and cognitive deficit mice. Asian Pac. J Trop. Biomed.. 2012;2(1):S104-S111.
- [Google Scholar]
- In silico study of some dexamethasone analogs and derivatives against SARs-CoV-2 target: a cost-effective alternative to remdesivir for various COVID phases. Curr. Chin. Sci.. 2022;2(4):294-309.
- [Google Scholar]
- Patel, K. B., Rajani, D., Ahmad, I., Patel, H., Patel, H. D., Kumari, P., 2023. Chrysin based pyrimidine-piperazine hybrids: design, synthesis, in vitro antimicrobial and in silico E. coli topoisomerase II DNA gyrase efficacy. Molecular Diversity, 1-16.
- Evaluation of oxindole derivatives as a potential anticancer agent against breast carcinoma cells: In vitro, in silico, and molecular docking study. Toxicol. In Vitro. 2023;86:105517
- [CrossRef] [Google Scholar]
- Mechanistic understanding from molecular dynamics in pharmaceutical research 2: lipid membrane in drug design. Pharmaceuticals. 2021;14(10):1062.
- [Google Scholar]
- Sasikumar, K., Ghosh, A. R., Dusthackeer, A., 2018. Antimycobacterial potentials of quercetin and rutin against Mycobacterium tuberculosis H37Rv. 3 Biotech, 8, 1-6.
- Preparation and evaluation of the ZnO NP–ampicillin/sulbactam nanoantibiotic: Optimization of formulation variables using RSM coupled GA method and antibacterial activities. Biomolecules. 2019;9(12):764.
- [Google Scholar]
- Shaw D. E. Research, Schrödinger Release (2020-3). Desmond molecular dynamics system. Maestro-Desmond interoperability tools.
- ATP-binding cassette (ABC) import systems of Mycobacterium tuberculosis: target for drug and vaccine development. Emerging Microbes Infect.. 2020;9(1):207-220.
- [Google Scholar]
- Silybin B and cianidanol inhibit Mpro and spike protein of SARS-CoV-2: Evidence from in silico molecular docking studies. Curr. Pharm. Des.. 2021;27(32):3476-3489.
- [Google Scholar]
- Structure based pharmacophore modelling approach for the design of azaindole derivatives as DprE1 inhibitors for tuberculosis. J. Mol. Graph. Modell.. 2020;101:107718
- [Google Scholar]
- Profiling the Structural determinants of pyrrolidine derivative as gelatinases (MMP-2 and MMP-9) inhibitors using in silico approaches. Comput. Biol. Chem.. 2023;107855
- [CrossRef] [Google Scholar]
- Challenges in drug discovery for intracellular bacteria. Pathogens. 2021;10(9):1172.
- [Google Scholar]




