

Translate this page into:
Evaluation of potential substrates and inhibitors of MRP2 transporter to predict effective combinatorial chemotherapeutic agents for treating MRP2-associated non-responsive colorectal cancer
*Corresponding author: E-mail address: fopo@salud.unm.edu (Md. F.A.D. Opo)
-
Received: ,
Accepted: ,
Abstract
Colorectal cancer (CRC) is a significant cause of death globally, due to the emergence of multidrug resistance (MDR), which limits the effectiveness of conventional chemotherapy. Multidrug resistance-associated protein 2 (MRP2) plays a critical role in the drug resistance observed in cancer. MRP2 contributes to cross-resistance to several structurally and functionally diverse chemotherapeutic drugs. This study aims to evaluate potential anticancer agents and inhibitors of MRP2 to develop effective therapeutic strategies for MRP2-associated non-responsive CRC. In this study, molecular docking was performed to reveal the MRP2 binding sites and affinity with anticancer drugs. Interaction analysis of chemotherapeutic drugs with MRP2 demonstrated irinotecan>doxorubicin>capecitabine>trifluridine>oxaliplatin>gemcitabine>tipiracil>5-Fluorouracil (5-FU) to be the decreasing order of binding affinities. 5-FU exhibited the lowest binding affinity, while irinotecan displayed the highest. In contrast, docking analysis of inhibitors with MRP2 showed probencid<MK-571<S-(2,4-dinitrophenyl) glutathione<dihydromyricetin <zafirlukast< montelukast to be the order of increasing binding affinities. Montelukast showed the highest binding affinity with MRP2. Notably, our findings showed that irinotecan, oxaliplatin, montelukast, and zafirlukast bind specifically to MRP2 regions TM12 and TM15. Our results suggest that 5-FU could be a more effective option for MRP2-overexpressing CRC as it interacts poorly with MRP2. Additionally, gemcitabine and oxaliplatin shared common binding sites, implying that competitive binding may help overcome MDR. Furthermore, our findings imply that a combinatorial approach utilizing irinotecan/oxaliplatin and an inhibitor may offer an efficient approach to combat drug resistance in CRC, paving the way for improved patient outcomes.
Keywords
ATP-binding cassette (ABC)
Chemotherapy
Colorectal cancer
Inhibitors
MRP2
Multidrug resistance

1. Introduction
Colorectal cancer (CRC) ranks as the second most lethal tumor worldwide recording more than 0.9 million deaths in 2020 (Luo et al., 2021; Siegel et al., 2023). Substantial progress has been made in the development of new classes of chemotherapeutic agents to treat CRC over the past decades, such as bevacizumab, oxaliplatin, pembrolizumab, 5-fluorouracil (5-FU) and irinotecan. Nonetheless, the prognosis for CRC remains poor with a 5-year survival rate of around 64–67%. This trend seems to decrease to 14–15% in patients with distant cancer (Mattiuzzi et al., 2019). Several studies have indicated that multidrug resistance (MDR) remains a major challenge in CRC treatment, resulting in poor prognosis (Ahluwalia et al., 2021; Chen et al., 2022). Therefore, understanding the MDR mechanism and devising new strategies to hinder or overcome drug resistance are prerequisites for improving the treatment outcomes. The mechanisms involved in driving resistance are complex and have not been well established (Marusyk et al., 2020). However, it is well understood that drug resistance arises through several mechanisms, including inhibition of drug uptake, overexpression of drug transporters, target-associated signaling, escape from the immune response, and the emergence of drug-tolerant cells. Notably, among the different mechanisms, the overexpression of ABC transporters belonging to the ATP binding cassette (ABC) family is considered the most prevalent in conferring MDR in cancer cells (Haimeur et al., 2004; Luo et al., 2021; Poku and Iram, 2024). Among these ABC transporters, multidrug resistance-associated protein 2 (MRP2) has been widely studied with reference to the gastrointestinal tissue where MRP2 is expressed differentially and engaged chiefly in exporting numerous substrates from the epithelium into the luminal side of the intestine. Thus, the MRP2 pump appears to be essential for transporting harmful substances such as toxins, metabolites, waste, and carcinogenic compounds as a substrate from intestinal cells to protect the body from exposure to toxic and lethal effects (Marin et al., 2020). The efflux pump MRP2 is abundantly observed on the apical membrane (canalicular domain), a narrow lumen connecting the two adjacent hepatocytes, epithelial cells of the renal proximal tubule, and the apical membrane of enterocytes of the duodenum and jejunum (Karpen and Karpen, 2017). MRP2 mediates the exports of several metabolites, conjugated and unconjugated, and anionic drugs including estradiol-17beta-glucuronide, estrone sulfate as well as bilirubin glucuronide and thus bilirubin transport by MRP2 is the main mechanism for bile acid–independent bile flow across the canaliculus (Heredi-Szabo et al., 2008; Järvinen et al., 2018; Keppler, 2014). MRP2 dysfunction could lead to conditions like Dubin-Johnson syndrome, most often characterized by increased levels of bilirubin glucuronide in the bloodstream resulting in jaundice (Deng et al., 2020; Mazza et al., 2024). Notably, MRP2 has also been reported to be highly expressed in colon adenomas and carcinomas as compared to normal tissue (Andersen et al., 2015) and this differential expression level indicates that it could be afflicted with an early event in the onset and development of CRC (Andersen et al., 2015; Marin et al., 2020). It has been well-documented that MRP2 overexpression facilitates the efflux of chemotherapeutic drugs from cancer cells, resulting in resistance to anticancer agents (Sticova et al., 2013; Sui et al., 2016). Notably, MRP2 has also been reported to be expressed in distinct cisplatin (CDDP)-resistant cancer cell lines, including colon cancer, ovarian cancer, bladder cancer, and hepatocellular carcinoma (Liedert et al., 2003; Materna et al., 2005). In vitro studies have also confirmed the implication of MRP2 in the MDR mechanism, as it is characterized by the failure of chemotherapeutic agents to sensitize many cancer cell lines, including CRC (Ma et al., 2009; Materna et al., 2006). Other studies have also established that the overexpression of MRP2 in cancer cells leads to the efflux of several structurally distinct chemotherapeutic agents, including cisplatin, irinotecan, paclitaxel, methotrexate, oxaliplatin, and/or vincristine, along with glutathione-conjugated drugs (Maarten T Huisman et al., 2005; Myint et al., 2019, 2015a). A study conducted by Korita et. al. demonstrated that the expression level of MRP2 is a key determinant in establishing the efficacy of cisplatin-based chemotherapy of hepatocellular carcinoma. (Korita V. et al., 2010). Furthermore, a recent in vitro study by Biswas et. al. showed that oxaliplatin is a substrate of MRP2 with possibly two binding sites (Biswas et al., 2019). They demonstrated that silencing MRP2 leads to an increase in oxaliplatin accumulation and cytotoxicity in gastrointestinal cell lines such as CRC cells (Caco-2) and pancreatic cancer cells (PANC-1) (Biswas et al., 2019).
Hence, it is well understood from numerous studies that overexpression of MRP2 is the leading cause of efflux of therapeutic drugs, and as a result, the MRP2 transporter is considered one of the major contributors to chemotherapeutic resistance in CRC (Sticova et al., 2013; Sui et al., 2016). From this perspective, revealing the structure and function of MRP2 transport protein has become imperative in order to recognize its drug-binding site through which the MRP2 pump recognizes drugs as a substrate and effluxes the drugs out of the tumor cell. It is also important to calculate its binding affinities with such drugs. This is an important aspect in devising better strategies and treatment modalities to overcome MDR in patients with cancer. Moreover, analysis and validation of drugs that could potentially block the MRP2 transporter through competitive inhibition within the pump or inhibit MRP2 expression or function have drawn significant attention toward improving the efficacy of chemotherapeutic agents in cancer patients.
Based on ABC transport protein structure and function prediction, the MRP2 is composed of a larger core segment with two transmembrane domains (TMDs), two nucleotide-binding domains (NBDs), and a linker segment L1 (Borst and Elferink, 2002). Furthermore, MRP2 contains a third NH2-terminal membrane-spanning domain, MSD0, also called the terminal transmembrane domain (Borst and Elferink, 2002; Kruh and Belinsky, 2003). Functional prediction of MRP2 revealed that TM helices contain basic residues that are involved in the recognition and translocation of their substrates (Ryu et al., 2000). It has been reported that these basic residues in TM6, TM9, TM16, and TM17 are implicated in the binding of glutathione-conjugated drugs, and interestingly, a critical amino acid TM11 encourages steady expression of the MRP2 transporter (Ryu et al., 2000). Furthermore, an aromatic amino acid (Trp1254) residue present in TM17 is considered a key determinant in facilitating the transport of methotrexate by the MRP2 pump (Ito et al., 2001). Notably, according to transport assay vesicle analysis performed in the presence of various drugs, it has been reported that the MRP2 possibly contains distinct binding sites: at least one for drug transport and a second essentially associated with allosteric regulation of the first; depending on receiving response patterns it could be either complex inhibition or stimulation (Zelcer et al., 2003). On the other hand, several mutational analyses of model transport Pgp1 have revealed several critical residues in the TMD, loops, and NBD of MDR1 pumps involved in substrate binding, translocation, and efflux (Loo and Clarke, 2002; Tangella et al., 2020). Similarly, we also anticipate that MRP2 may have a wide range of key residues spanning TMD, loops, and NBD essentially involved in substrate recognition, translocation, and extrusion in this transporter.
There is currently limited information on how anticancer drugs with MRP2 transporter impact chemotherapeutic effectiveness. MRP2 protein, along with its pharmacokinetics and substrate specificity are poorly studied through in-silico investigations. A deeper understanding of MRP2 substrate specificities is crucial for selecting effective drugs and designing new chemotherapeutics to minimize MRP2-related drug interactions. Consequently, analyzing MRP2-substrate interactions to determine protein-ligand binding affinity and specificity is essential for predicting the effectiveness of anticancer drugs, especially in CRC patients with MDR.
Hence, in this study, we aimed to identify the binding site of MRP2 for several chemotherapeutic drugs and inhibitors in silico. Molecular docking analysis can demonstrate the binding affinity of drugs and identify potential interacting sites between MRP2 and its substrates. This approach may predict the most effective anticancer drugs as a combinatorial therapeutic strategy for treating non-responder CRC patients, often associated with overexpressed MRP2 transporters.
2. Materials and methods
2.1 Ligand preparation
The three-dimensional (3D) structure of the drugs was retrieved from the NCBI PubChem database in SDF format; 5-Fluorouracil (PubChem ID: 3385), Capecitabine (PubChem ID: 60953), Dihydromyricetin (PubChem ID: 161557), Doxorubicin (PubChem ID: 31703), Estradiol-17-beta-glucuronide (PubChem ID: 5281887), Estrone sulfate (PubChem ID: 3001028), Gemcitabine (PubChem ID: 60750), Irinotecan (PubChem ID: 60838), MK-571 (PubChem ID: 5281888), Montelukast (PubChem ID: 5281040), Oxaliplatin (PubChem ID: 9887053), Probenecid (PubChem ID: 4911), S-(2,4-dinitrophenyl) Glutathione (PubChem ID: 97535), SN-38 (PubChem ID: 104842), Tipiracil (PubChem ID: 6323266), Trifluridine (PubChem ID: 6256), and Zafirlukast (PubChem ID: 5717) (https://pubchem.ncbi.nlm.nih.gov/) (Kim et al., 2019; Wang et al., 2009). The ligands were prepared using Open Babel (O’Boyle et al., 2011). The structures were energy-minimized and converted to the .pdbqt (protein data bank, partial charge (Q), and atom type (T)) format using Open Babel integrated in the PyRx Tool v.0.8 (Dallakyan and Olson, 2015).
2.2 Receptor preparation
The human ABCC2 (MRP2) protein 3D structure (8JX7) was retrieved from the Protein Data Bank (PDB) (Berman et al., 2000). The structure was prepared by removing water molecules, adding missing hydrogens, assigning Kollman charges, and then converted to .pdbqt format using the AutoDock Tool in PyRx. The protein-ligand binding sites were predicted using P2RANKWeb 3 tool (Krivák and Hoksza, 2015) and the grid box was centered to include all pockets showing high scores.
2.3 Molecular docking
Molecular docking is a computational approach that predicts the most favorable orientation and binding affinity of a small molecule (ligand) to a target protein, forming a stable complex. It is a key tool in drug discovery, used to screen drug candidates and analyze their interactions with target proteins (Morris and Lim-Wilby, 2008). In the current study molecular docking was performed using PyRX version 0.8, with the AutoDock Vina algorithm. The exhaustiveness parameter was set to value 8, and the number of output poses was set to 10. Docking was performed using the prepared ligand and receptor files. The grid box parameters were identical to those used for receptor preparation.
2.4 Docking results analysis
The resulting docking poses were ranked based on their Vina scoring function. The top pose of each ligand was analyzed and visualized using BIOVIA, Dassault Systèmes, Discovery Studio Visualizer, v24.1.0.23298, San Diego: Dassault Systèmes, 2023. Key interactions, such as hydrogen bonds, hydrophobic contacts, and pi-pi stacking, were analyzed and documented (SYSTÈMES, n.d.).
3. Results
3.1 Analysis of docking results for anti-cancer drugs
Prior to molecular docking, we identified the binding pockets of MRP2 using the P2RANK web tool and found around 25 binding pockets. Based on the binding score, we used the top five binding pockets for docking, as presented in Table 1. Fig. 1 shows the binding pockets of MRP2. The docking analysis results for each drug are summarized in Table 1, detailing binding affinities, binding sites, and the number of hydrogen bonds. We performed docking of SN-38, estradiol, and estrone sulfate with MRP2 because these drugs are the known substrate of MRP2. We observed the highest binding affinity of SN-38 (-9.1), Estradiol-17-beta-glucuronide (-8.9 kcal/mol), and estrone sulfate (-8.2 kcal/mol), respectively, as shown in Table 2.
| Rank | Score | Probability | Number of residues |
|---|---|---|---|
| 1 | 14.03 | 0.722 | 16 |
| 2 | 13.33 | 0.702 | 27 |
| 3 | 12.15 | 0.663 | 14 |
| 4 | 9.928 | 0.542 | 27 |
| 5 | 5.88 | 0.308 | 17 |
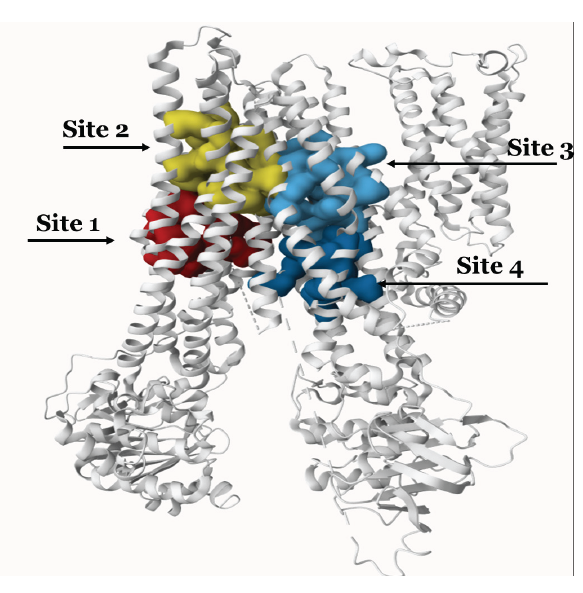
- Shows the binding pocket of MRP2.
| Drugs | Binding affinity (kcal/mol) | Hydrogen bonds | Other interactions |
|---|---|---|---|
| Known substrates | |||
| SN-38 | -9.1 | Asp884, Ser908 | Val421 (pi-alkyl), Arg911 (pi-cation), Tyr1160 (pi-pi) |
| Estradiol-17-beta-glucuronide | -8.9 | Asp884, Asn907, Gly1086 | Met492, Lys495, Leu499 (Alkyl, pi-alkyl) |
| Estrone sulfate | -8.2 | Lys495, Asp1093 | Leu499, Arg911 (Alkyl, pi-alkyl) Lys495 (pi-cation) |
| Anticancer drugs | |||
| Drugs | Binding affinity (kcal/mol) | Hydrogen bonds | Other interactions |
| Irinotecan | -9.7 | Thr912, Arg1156, Ser1164 | Gly418 (CHB), Val421 (Alkyl), Thr1160 (pi-pi) |
| Doxorubicin | -8.7 | Gln429, Ser908, Thr912, Ser1153 | Ser425 (CHB), Val421 (pi-alkyl), Tyr1160 (amide-pi-stacked) |
| Capecitabine | -6.8 | Arg911, Gly1086, Ser1089, Thr1264, Arg1271 | Leu499, Ala1085 (Alkyl), Asp884 (Halogen) |
| Trifluridine | -6.6 | Ile710, Asn712, Glu756, Ser1168 | Gly755 (CHB), Gln711, Glu1165 (Halogen) |
| Oxaliplatin | -6.4 | Gln429, Thr912, Arg1156, Tyr1160 | |
| Gemcitabine | -6.2 | Gln429, Asp884, Arg911, Thr912 | Ser425 (CHB, Halogen), Arg911 (pi-cation) |
| Tipiracil | -6.2 | Ser708, Ile710, Asn712, Glu1165 | Glu756 (pi-anion) |
| 5-Fluorouracil (5-FU) | -5.6 | Gln488, Asn491, Lys495, Glu532, Asp1093 | Phe539 (pi-pi), Leu536 (pi-alkyl), Asp1093 (pi-anion) |
3.1.1 Irinotecan
Our results revealed that Irinotecan (-9.7 kcal/mol) has the highest binding affinity to MRP2 with various interactions, including hydrophobic interactions and hydrogen bonding with Irinotecan and amino acids in the protein’s binding pocket shown in Fig. 2(a). The irinotecan forms three hydrogen bonds with Thr912, Arg1156, and Ser1164 residues. Additionally, weaker van der Waals interactions were observed with Leu913, Ser425, and Asn422 residues, further stabilizing the ligand as shown in Fig. 2(a). Hydrophobic interactions, including alkyl interactions with Val421 and Tyr1160 and Pi-Pi T-shaped interactions, enhance binding by positioning the ligand in non-polar regions of the protein. Most interactions, particularly the hydrophobic and hydrogen bonds, play a critical role in securing Irinotecan within the protein’s active site, ensuring its effective binding and potential activity.
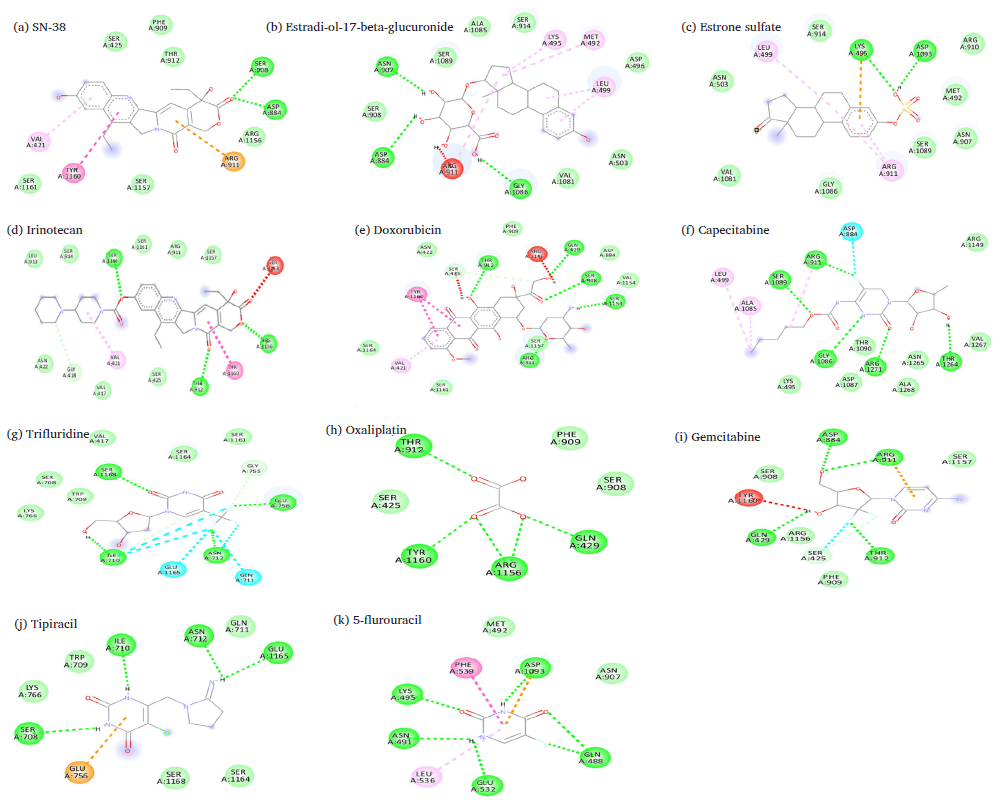
- Predicated binding sites of MRP2 with (a) irinotecan, (b) SN-38, (c) estradiol, (d) doxorubicin, (e) estrone sulfate, (f) capecitabine, (g) trifluridine, (h) oxaliplatin, (i) gemcitabine, (j) tipiracil, (k) 5-FU (Legends: Green: Van der Waals interactions, Green dashed-lines: Hydrogen bond, Gray dashed-lines: Carbon hydrogen bond, Cyan: Halogen interactions, Orange dashed-lines: Pi-cation interactions; Pink dashed-lines: Pi-Alkyl interactions (Pi-Pi stacked interactions); Light pink dashed-lines: Alkyl interactions; Red dashed-lines: unfavorable interactions.
3.1.2 SN-38
SN-38 (-9.1 kcal/mol), a derivative of Irinotecan, interacted with the MRP2 binding pocket in ways that contribute to stabilizing the ligand. Hydrogen bonds play a critical role, as Asp884 and Ser908 form strong bonds with the ligand, demonstrated in Fig. 2(b) and Table 1. These hydrogen bonds help secure the ligand firmly within the binding site. Pi-cation interactions, particularly with Arg911, further stabilize the ligand by involving the arginine’s positive charge with the ligand’s aromatic system. This interaction is important for anchoring the ligand in the pocket. Additionally, Pi-Pi T-shaped interactions are observed with Tyr1160 and Val421, helping to stabilize the ligand’s ring system by stacking it with the aromatic groups of the protein. The van der Waals forces between the ligand and Phe909, Ser425, and Thr912 residues add stability by providing gentle interactions that keep the ligand in position.
3.1.3 Estradiol-17-beta-glucuronide
Estradiol-17-beta-glucuronide (-8.9 kcal/mol) shows several interactions with the amino acid residues of MRP2, as shown in Table 1, including three hydrogen bonds that are crucial for binding, with Arg911, Gly1086, and Asn907 forming strong stabilizing bonds as shown in Fig. 2(c). These interactions help secure the ligand in an optimal position. Additionally, there are several van der Waals interactions with Ser908, Ala1085, Ser1089, Ser914, Asn503, val1081, and Asp496 residues, providing weaker yet supportive interactions that contribute to the overall stability of the ligand. The ligand also experiences alkyl and pi-alkyl interactions with hydrophobic residues like Lys495, Leu499, and Met492, which help anchor the non-polar regions of the ligand within the binding pocket. These interactions are particularly important for stabilizing hydrophobic portions of the ligand. Estradiol-17-beta-glucuronide is held in place through strong hydrogen bonds, weaker van der Waals forces, and hydrophobic interactions.
3.1.4 Doxorubicin
Doxorubicin (-8.7 kcal/mol) shows various interactions with amino acid residues of the MRP2 binding site, as shown in Fig. 2(d). Four hydrogen bonds with Gln429, Ser904, Thr912, and Ser1153 are crucial for forming strong stabilizing bonds with Doxorubicin. These interactions help secure the ligand in an optimal position. Additionally, there are several van der Waals interactions with Ser908, Ala1085, and Asp884 residues, providing weaker yet supportive contacts that contribute to the structural integrity of the ligand. The ligand also formed Pi-Alkyl interactions with hydrophobic residues like Va421, which help anchor the non-polar regions of the ligand within the binding pocket. These interactions are particularly important for stabilizing the hydrophobic portions of the ligand. Additionally, we observed a carbon-hydrogen bond at residue Ser425 and amide pi-stacked interaction at Tyr1160, as shown in Table 2.
3.1.5 Estrone sulfate
We have observed that Estrone sulfate (-8.2 kcal/mol) formed two hydrogen bonds with Asp1093 and Arg911 in the MRP2 binding sites, as shown in Table 2 and Fig. 2(e). pi-cation interactions with Lys495 further strengthen the binding by stabilizing the aromatic ring of Estrone sulfate. Alkyl and pi-alkyl interactions with residues like Leu499 and A911 contribute to hydrophobic stabilization, helping to anchor non-polar regions of the ligand. Additional van der Waals interactions with Ser914 and Met492 residues are weaker but supportive in keeping the ligand in place.
3.1.6 Capecitabine
Capecitabine (-6.8 kcal/mol) formed five hydrogen bonds with Arg911, Gly1086, Ser1089, Thr1264, and Arg1271 residues, which play an essential role in anchoring the ligand, as presented in Fig. 2(f) and Table 2. Additionally, halogen bonds with Asp884 enhance stability by interacting with the fluorine atom of capecitabine. Alkyl interactions with Leu499 and Ala1085 provide hydrophobic stabilization, while van der Waals interactions with Lys495 and Val1267 are supportive but weaker interactions that help hold the ligand in place.
3.1.7 Trifluridine
Fig. 2(g) represents that Trifluridine (-6.6 kcal/mol) forms four hydrogen bonds with residues, including Ile710, Asn712, Glu756, and Ser1168, which help to hold it in place securely. It also forms halogen bonds between its fluorine atoms and residues Asn711 and Gln711, adding to its stability. There are also van der Waals interactions with Ser1168 and Glu1165, which are weaker but still help keep the molecule in the right position. Carbon-hydrogen bonds with Gly755 add more stability, as shown in Table 2.
3.1.8 Oxaliplatin
Oxaliplatin (-6.4 kcal/mol) made four hydrogen bonds with Thr912, Tyr1160, Arg1156, and Gln429, providing strong stabilizing forces that anchor the ligand, as demonstrated in Fig. 2(h) and Table 2. While van der Waals interactions with Phe909, Ser908, and Ser425 were weaker but supportive, a network of hydrogen bonds plays the dominant role in securing oxaliplatin within the active site, ensuring its effective positioning for potential biological activity.
3.1.9 Gemcitabine
Gemcitabine (-6.2 kcal/mol) highlights the various interactions that stabilize the ligand within the MRP2 binding site. Gemcitabine interacts with MRP2 though four hydrogen bonds are formed with Gln429, Asp884, Arg911, and Thr912, shown in Fig. 2(i). Additionally, pi-cation interaction with Arg911, further strengthens the ligand’s position. Halogen bonds with Thr912 provide additional stability in the presence of fluorine atoms in the ligand. Van der Waals interactions with residues like Phe909 and Ser908 are weaker but supportive.
3.1.10 Tipiracil
Fig. 2(j) shows that tipiracil (-6.2 kcal/mol) formed four hydrogen bonds with Asn712, Ile710, Gln711, and Glu1165. Additionally, a pi-anion interaction with the aromatic system of Glu756 helps further stabilize the ligand, as depicted in Table 2. Van der Waals interactions with residues such as Ser708, Trp709, and Lys766 provide additional, weaker support, ensuring the ligand remains in place. Together, these interactions ensure that tipiracil is effectively bound within the MRP2, facilitating its biological function.
3.1.11 5-Fluorouracil (5-FU)
5-FU (-5.6 kcal/mol) showed the lowest binding affinity with MRP2, as presented in Table 2. 5-FU formed five hydrogen bonds with Lys495, Asp1093, Asn491, Asp1093, and Gln488 of MRP2. Additionally, pi-anion interactions with Asp1093 contribute to stabilizing the negatively charged regions of the ligand. Pi-Pi Stacked interactions with Phe539 and Pi-Alkyl interactions with Leu536 and Asn491 help to stabilize the aromatic and hydrophobic regions of 5-FU, as shown in Fig. 2(k). Van der Waals interactions with residues like Met492 and Asn907 are weaker but supportive.
3.2 Analysis of docking results for inhibitors
3.2.1 Montelukast
The docking analysis of Montelukast (-8.8 kcal/mol) reveals that it formed five hydrogen bonds with Ser908, Thr912, Arg911, Ser1153, and Asp884 residues, which help it to anchor firmly within the binding pocket as shown in Fig. 3(a) and Table 3. Additionally, there are pi-cation interactions involving Arg1156, where the positively charged arginine side chain interacts with the aromatic rings of the ligand, further stabilizing its position. The ligand also shows van der Waals interactions with residues, which provide weaker but essential contacts to help secure Montelukast in the pocket. Moreover, pi-alkyl interactions with Arg1149, Arg1150, Tyr1160, and Phe909 create hydrophobic interactions that stabilize the non-polar parts of the ligand.
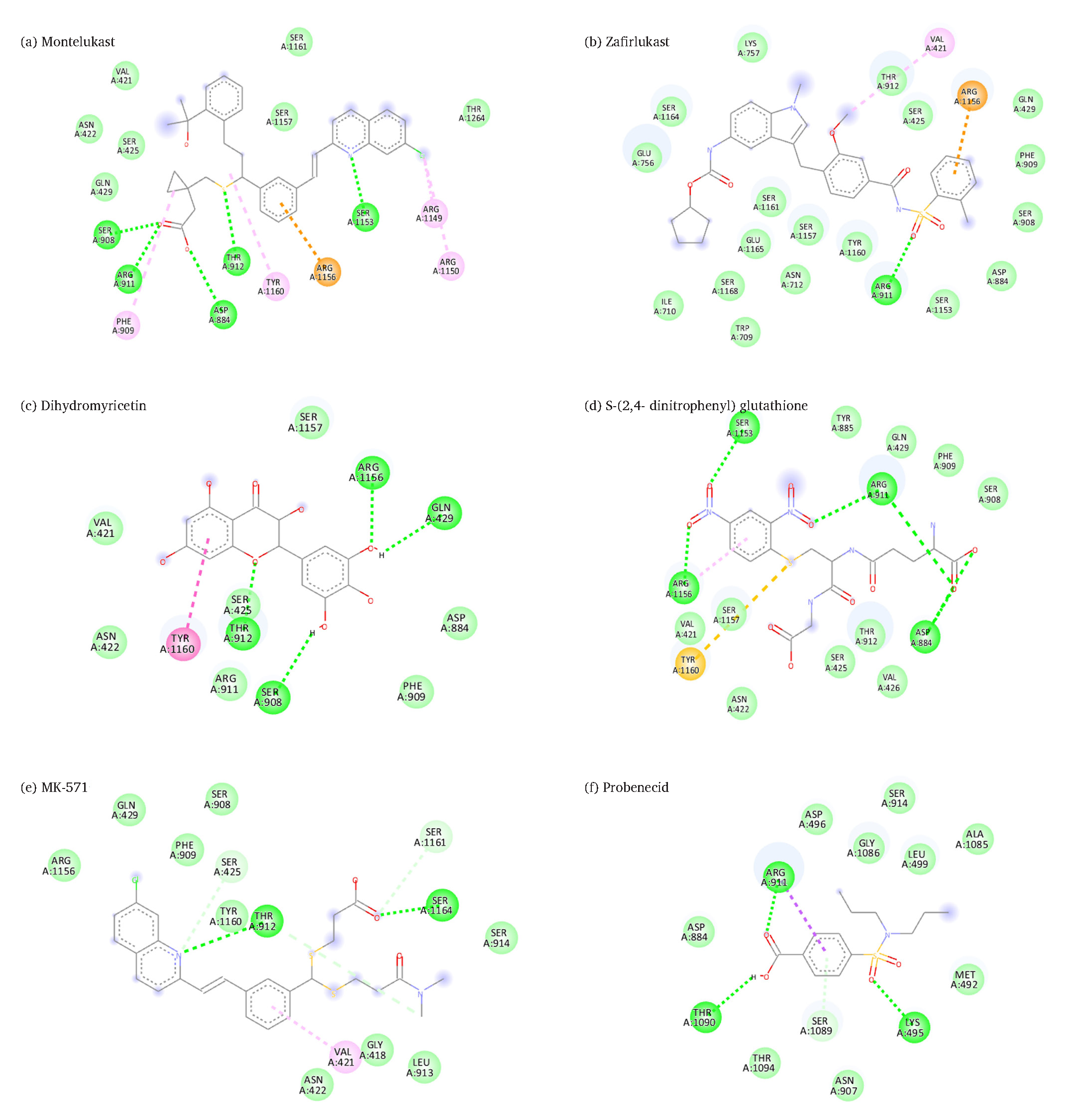
- Predicated binding sites of MRP2 with (a) montelukast, (b) zafirlukast, (c) dihydromyricetin, (d) S-(2,4- dinitrophenyl) glutathione, (e) MK-571, (f) probenecid, (Legends: Green: Van der Waals interactions, Green dashed-lines: Hydrogen bond, Gray dashed-lines: Carbon hydrogen bond, Cyan: Halogen interactions, Orange dashed-lines: Pi-cation interactions; pink dashed-lines: Pi-Alkyl interactions (Pi-Pi stacked interactions); light pink dashed-lines: alkyl interactions; red dashed-lines: unfavorable interactions.
| Inhibitor | Binding affinity (kcal/mol) | Hydrogen bonds | Other interactions |
|---|---|---|---|
| Montelukast | -8.8 | Asp884, Ser908, Arg911, Thr912, Ser1153 | Phe909, Arg1149, Arg1150, Tyr1160 (Alkyl, pi-alkyl) Arg1156 (pi-cation) |
| Zafirlukast | -8.7 | Arg911 | Val421 (Alkyl), Arg1156 (pi-cation) |
| Dihydromyricetin | -7.5 | Gln429, Ser908, Thr912, Arg1156 | Tyr1160 (pi-pi) |
| S-(2,4-dinitrophenyl) glutathione | -7.2 | Asp884, Arg911, Ser1153, Arg1156 | Arg1156 (pi-Alkyl), Tyr1160 (pi-sulfur) |
| MK-571 | -7.1 | Thr912, Ser1164 | Val421 (pi-alkyl) Ser425, Ser1161 (CHB) |
| Probenecid | -6.1 | Lys495, Arg911, Thr1090 | Arg911 (pi-sigma), Ser911 (pi-sigma) |
3.2.2 Zafirlukast
Docking results showed that Zafirlukast (-8.7 kcal/mol) formed only single hydrogen bonds with residue Arg911, which may play a significant role in anchoring the ligand. Additionally, van der Waals interactions with multiple residues provide further stability by gently holding the ligand in place, as shown in Fig. 3(b). A pi-cation interaction with Arg1156 strengthens the binding by leveraging electrostatic forces between the positively charged arginine and the aromatic ring of Zafirlukast. Alkyl interactions with Val421 contribute to stabilizing the hydrophobic regions of the ligand, as demonstrated in Table 3.
3.2.3 Dihydromyricetin
Dihydromyricetin (-7.5 kcal/mol) formed four hydrogen bonds with Arg1156, Gln429, Thr912, and Ser908, as represented in Table 3. Additional hydrogen bonds with Thr912 and Ser425 further strengthened the ligand’s positioning. Van der Waals interactions with residues, are supportive but weaker and help maintain the ligand’s stability. A Pi-Pi T-shaped interaction with Tyr1160 stabilizes the ligand’s aromatic system, as shown in Fig. 3(c).
3.2.4 S-(2,4-dinitrophenyl) glutathione
S-(2,4-dinitrophenyl) glutathione (-7.2 kcal/mol) formed four hydrogen bonds between Arg911, Asp884, Arg1156, and Ser1153, which anchor the ligand firmly, as directed in Table 3. Pi-sulfur interactions between Tyr1160 and the sulfur atom of the ligand create additional stability. In contrast, pi-alkyl interactions with Arg1156 help secure the non-polar regions of the molecule. Van der Waals forces with surrounding residues such as Asn422, Val426, and Ser908 provide further support by gently holding the ligand in place, as shown in Fig. 3(d).
3.2.5 MK-571
Our docking results show that MK-571 (-7.1 kcal/mol) formed two hydrogen bonds with residues Thr912 and Ser1164, as shown in Figs. 3(e). Two carbon-hydrogen bonds with residues like Ser425 and Ser1161 further stabilize the molecule. Pi-alkyl interactions with Val421 and Asn422 provide additional stabilization to the hydrophobic regions of the ligand. Weak but supportive van der Waals interactions gently hold the ligand in place, as shown in Fig. 3(e).
3.2.6 Probenecid
Probenecid (-6.1 kcal/mol) formed two hydrogen bonds with Arg911 and Lys495, which help anchor the ligand, as depicted in Table 2. Additionally, pi-sigma interactions with Arg911 stabilize the aromatic ring of probenecid, while pi-donor hydrogen bonds with Ser1089 contribute to further stability. Van der Waals interactions provide weaker but essential contacts to maintain the ligand’s position, as shown in Fig. 3(f).
4. Discussion
The development of MDR and relapse of tumor cells that repeatedly occurs during cancer therapy is a key limitation in the successful treatment of cancer, even after using adequate doses of chemotherapeutic agents to eliminate cancer cells. (Ashique et al., 2024; Mirakhorli et al., 2012). Currently, platinum-based oxaliplatin is routinely used in the treatment of CRC but using a combination of oxaliplatin and 5-FU has shown an improvement in the efficacy of adjuvant chemotherapy, especially in case of relapses (Casado et al., 2007). Nevertheless, certain patients with CRC continue as non-responders to effective combinatorial chemotherapeutic approaches (Arts et al., 1999; Casado et al., 2007). In this perspective, there is a need to investigate the interaction of anticancer drugs with the MRP2 transporter to predict the nature of the drug binding site and its affinity potential with which the MRP2 pump recognizes chemotherapeutic drugs as a substrate for efflux.
In the present study, we performed molecular docking with its three known substrates SN-38, Estradiol-17-beta-glucuronide, and estrone sulfate (Herédi-Szabó et al., 2009; Zhang et al., 2022). We found high binding affinities of SN-38 (-9.1 kcal/mol), Estradiol-17-beta-glucuronide (-8.9 kcal/mol), and estrone sulfate (-8.2 kcal/mol) as shown in Table 2. Estradiol-17-beta-glucuronide is an estrogen metabolite produced in the liver and later excreted in the bile. Estradiol-17-beta-glucuronide is a well-known substrate of MRP2, and serves as a cholestatic agent that reduces bile flow (Gerk et al., 2004; Herédi-Szabó et al., 2009). The functional role of hepatic MRP2 is directing bile salts, as well as glucuronide and sulfate conjugates, including estrone sulfate and glutathione, to bile (Järvinen et al., 2018; Loe et al., 1996). The interaction studies between MRP2 and its three known substrates (SN-38, Estradiol-17-beta-glucuronide, and estrone sulfate) helped establish a basis for comparing anticancer drug interactions with MRP2. This comparison can predict which anticancer drugs may be more effective, based on the idea that drugs with lower binding affinity to MRP2, compared to natural substrates, could be more potent.
Furthermore, we evaluated the interaction of anti-cancer drugs with MRP2 to reveal the binding affinity and potential binding sites of MRP2. Most drug binding sites were present in TM8, TM9, TM10, TM12, TM14, and TM15, as shown in Fig. 4, while only trifluridine and tipiracil showed binding in NBD1. Our ligand–MRP2 protein alignment analysis revealed irinotecan binding to TMD12 and TM15, and the 5-FU binding site was found in TM9, TM10, and TM14 of MRP2, as shown in Fig. 4.
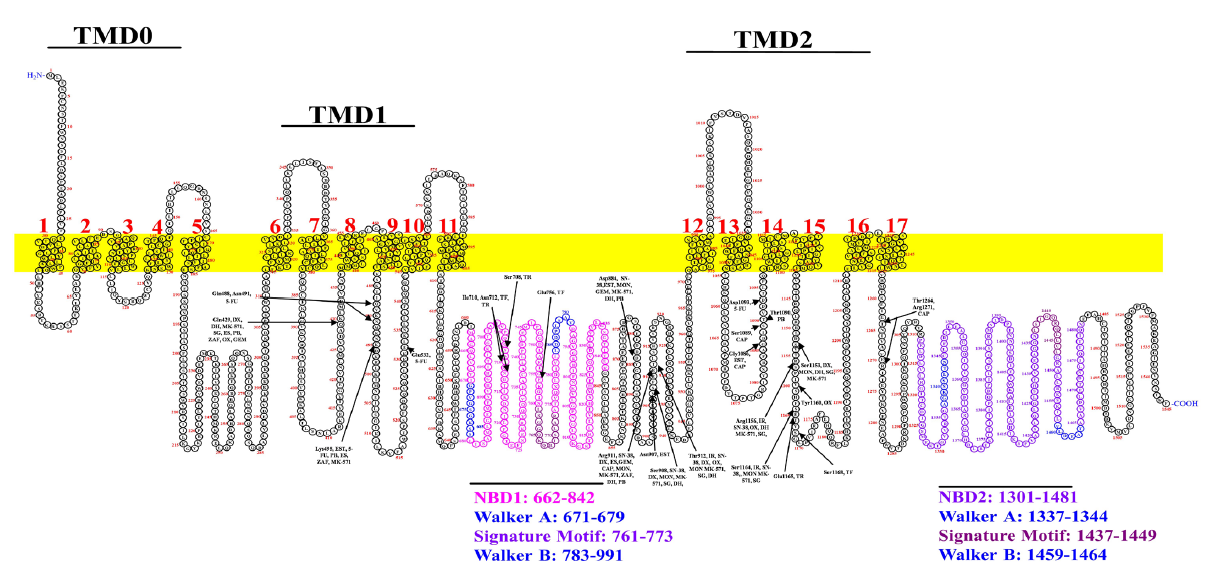
- Predicts the binding sites of anticancer drugs and inhibitors with MRP2. Drugs are represented with abbreviations; Irinotecan, IR; SN-38, Estradiol, EST; Doxorubicin, DX; Estrone sulfate, ES; Capecitabine, CAP; Trifluridine, TF; Oxaliplatin, OX; Gemcitabine, GEM; Tipiracil, TR; 5-Fluorouracil, 5-FU; Monte-lukas, MON;, Zafirlukast, ZAF; Dihydromyricetin, DH; S-(2,4-dinitrophenyl) glutathione, SG; MK-571; and Probenecid, PB.The NBD1 & NBD2 and conserved regions are depicted in distinct colors.
Molecular docking analysis revealed that irinotecan (-9.7 kcal/mol) was found to have the highest binding affinity for MRP2. Notably, irinotecan formed three hydrogen bonds with MRP2 at residues Thr912, Arg1156, and Ser1164. Additionally, it formed a carbon-hydrogen bond, alkyl, and pi-pi interactions at residues Gly418, Val421, and Thr1160 of MRP2, respectively, as shown in Fig. 2(a). Our analysis suggests that irinotecan is likely the best substrate, as shown in Table 2. Hence, our findings indicate that irinotecan interacts with the highest binding affinity for MRP2 and, thus, may not be considered a better option for overexpressed MRP2 in CRC tumor cells. The docking analysis also showed doxorubicin (-8.7 kcal/mol), capecitabine (-6.8 kcal/mol), trifluridine (-6.6 kcal/mol), oxaliplatin (-6.4 kcal/mol), gemcitabine (-6.2 kcal/mol), tipiracil (-6.2 kcal/mol), and 5-FU (-5.6 kcal/mol) ranked as second, third, fourth, fifth, sixth, seventh and eighth based on binding affinity in anticancer drugs with MRP2, as shown in Table 2. Our docking results showed that doxorubicin has the second-highest binding affinity, forming interactions with TM8, TM12, and TM15, as shown in Fig. 4. Our findings reveal that doxorubicin may act as the substrate of MRP2 due to its higher binding affinity. It is reported that doxorubicin is a commonly used anticancer drug to treat various cancers, including CRC (Xiong and Xiao, 2018). Still, the emergence of MDR limits its effectiveness in CRC (Khaleel et al., 2016), which is why it cannot be used in MDR-overexpressed CRC. Additionally, it was observed that capecitabine binds in the TM12, TM14, and TM17 of MRP2 regions, as shown in Fig. 4. Currently, capecitabine is used to treat advanced-stage metastatic CRC (Pasetto and Monfardini, n.d.; Pouya et al., 2023), but further studies are required to confirm its efficacy in MDR-overexpressed CRC patients. Our results showed that the trifluridine and tipiracil binding affinities were -6.6 kcal/mol and -6.2 kcal/mol, respectively, which implies that these may weakly bind with MRP2. Hence, it may be a viable choice for CRC treatment. Our results also showed that trifluridine and tipiracil share a common binding site in Ile710. Notably, these are the only anticancer drugs in the current study that bind in NBD1 of MRP2 protein, as shown in Fig. 4. It has been reported that trifluridine/tipiracil improves survival compared to palliatives. Currently, available therapies with a combination of trifluridine/tipiracil (Lonsurf) have been used as useful treatment options for the management of refractory CRC in patients. (Burness and Duggan, 2016; Patel et al., 2021). Lonsurf also overcomes MDR in human gastric 5-FU-refractory cells with elevated thymidylate synthase expression (Matsuoka et al., 2018).
Our result analysis showed that oxaliplatin interacts with the binding affinity of -6.4 kcal/mol through the formation of four hydrogen bonds at residues Gln429, Thr912, Arg1264, and Arg1160 of MRP2 protein, as shown in Table 2. We have observed that oxaliplatin binds in the TM8, TM12 and TM15 regions, as illustrated in Fig. 4. Previous studies have reported that the inhibition of MRP2 increases oxaliplatin accumulation and cytotoxicity in the colorectal cancer cell (Caco-2) and pancreatic cancer cell (PANC-1) and thus concluded that oxaliplatin may be a substrate of MRP2 with possibly two binding sites (Biswas et al., 2019). Thus, based on our findings, we suggest that oxaliplatin might not be alone considered an appropriate choice for the treatment of MRP2-overexpressed CRC patients. Additionally, Comella et al. (2009), reported that oxaliplatin in combination with 5-FU showed a 60% or higher response rate in the first line of treatment. Still, the response was recorded as approximately 25–50% for the relapse of CRC (Comella et al., 2009). Hence, we also suggest that treatment with oxaliplatin and 5-FU may be a suitable combinatorial approach to treat MRP2 overexpressed CRC as oxaliplatin alone can bind to MRP2 with high binding affinity as compared to 5-FU. Notably, gemcitabine showed one of the least binding affinity anticancer drugs, interacting through TM8 and TM15 of MRP2, as presented in Fig. 4. Studies have reported that gemcitabine is used in metastatic CRC patients (Baretti et al., 2021; Zeng et al., 2023). Thus, gemcitabine appeared to be a potent anticancer drug for MRP2-resistant CRC; however, this observation warrants further in vitro/in vivo studies to confirm its efficacy. Interestingly, our findings showed that 5-FU interacts with the lowest binding affinity (−5.6 kcal/mol), indicating it as a poor substrate of MRP2. Thus, it signifies a potent anticancer drug for CRC. Parallel studies have also suggested that 5-FU therapy significantly improves the survival in many cancers, including CRC, as evidenced by the fact that 5-FU continues to be the primary treatment option for CRC (Cho et al., 2020; Pardini et al., 2011). Hence, evidence supports our finding that 5-FU could be a potent drug option for CRC.
The MRP2 pump involves transporting various anticancer agents, including cisplatin, irinotecan, vincristine, and paclitaxel (Chu et al., 1997; Cui et al., 1999; Maarten T. Huisman et al., 2005). Other studies have also established that the overexpression of MRP2 in cancer cells leads to the efflux of several structurally distinct chemotherapeutic agents, including methotrexate, oxaliplatin, and/or vincristine, along with glutathione (Myint et al., 2015b). Furthermore, a study conducted by Korita et al. demonstrated that the expression level of MRP2 is a key determinant in establishing the effectiveness of cisplatin-based chemotherapy in hepatocellular carcinoma patients (Korita V. et al., 2010). Therefore, modulation of MRP2 expression or function through selective inhibitors is needed to improve the efficacy of chemotherapeutic drugs in CRC. A diverse range of structurally distinct drugs and compounds that may modulate or inhibit MRP2 expression or function has been identified. For example, the drug probenecid has been reported to treat gout disease, and another MK-571 is used as an antagonist of the leukotriene receptor to treat chronic asthmatic patients (Chen et al., 1999; Kim et al., 2001). Interestingly, Montelukast, a drug usually used in the treatment of asthma, has also been reported to inhibit the efflux of paclitaxel and saquinavir drugs in overexpressed MRP2 cells (Roy et al., 2009a). Therefore, we sought to analyze these drugs/inhibitor interactions with MRP2 to compare the binding affinity and binding sites. This comparison will help to determine whether the drugs function as modulators or inhibitors by competing with anticancer agents, potentially blocking MPR2 transport function. To shorten drug development time and increase the success rate of existing approved drugs in clinical trials, we aimed to analyze a few of these marketed drugs as inhibitors or modulators of MRP2 transport function. The rationale behind choosing approved drugs is that they have already been tested for safety and have better pharmacokinetic properties.
Our docking analysis of MRP2 with inhibitors indicated that Montelukast (-8.8 kcal/mol) interacts with the highest binding affinity with MRP2 as compared to Probenecid (-6.1 kcal/mol) which showed the least binding affinity to MRP2 as depicted in Table 2. Zafirlukast (-8.7 kcal/mol), Dihydromyricetin (-7.5 kcal/mol), S-(2,4-dinitrophenyl) glutathione (-7.2 kcal/mol), MK-571 (-7.1 kcal/mol) and Probenecid (-6.1 kcal/mol) ranked second, third, fourth, fifth and sixth, respectively, as shown in Table 3. Our findings suggested that Montelukast is the most effective inhibitor of MRP2 based on the highest binding affinity, and forms five hydrogen bonds with Asp884, Ser908, Arg911, and Thr912, which are present in TM12 and Ser1153 present in TM15 as shown in Fig. 3. Notably, we observed that Montelukast and zafirlukast formed the binding through a common interacting transmembrane at residue Arg911, i.e. TM 12 of MRP2, thus highlighting that TM12 may play a critical role in substrate recognition and binding as well as inhibition. Hence, we suggest that Montelukast could be utilized as a potent competitive inhibitor of MRP2 through selective inhibition of the key binding residues of TM11, essentially involved in substrate binding and transport. However, a previous comparison analysis among zafirlukast, MK-571, and Montelukast suggested that montelukast was the best inhibitor of MRP2 transport function (Roy et al., 2009a). This supports that montelukast could also be a good inhibitor of MRP2, as suggested by the current study. In addition, our docking findings suggest that Zafirlukast is the second-best inhibitor as it binds with the second-highest binding affinity of -8.7 kcal/mol to the MRP2 transporter. Moreover, we revealed that zafirlukast interacts with residues Arg911 of TM12 of MRP2, respectively, as demonstrated in Fig. 4. As it is well established that these trans membrane regions are critical in substrate recognition and binding, binding of zafirlukast to them could lead to a conformational change that possibly does not allow further binding of its substrates and leads to inhibition of drug transport (Ito et al., 2001; Ryu et al., 2000). Further, it has been well-known that TMDs are involved in encouraging the steady-state expression of MRP2. Hence, the binding of inhibitors may modulate the steady-state expression of MRP2 protein. As a result, lower expressions of MRP2 could significantly affect the efflux of anticancer agents (Ryu et al., 2000). Our findings suggested that Montelukast is a better inhibitor than Zafirlukast, which is in line with the previous findings (Roy et al., 2009b). Notably, our findings are validated by the above observation that Zafirlukast could also inhibit the MRP2 transport activity; however, further in vitro work may be required to establish whether Montelukast or Zafirlukast is a potential inhibitor of the MRP2 pump. Based on the binding affinity prediction, our findings revealed that dihydromyricetin (-7.5 kcal/mol) might be the third-best inhibitor of MRP2 after zafirlukast, as demonstrated in Table 3. Dihydromyricetin forms four hydrogen bonds with MRP2 at Gln429 (TM8), Ser908, Thr912 (TM12), and Arg1156 (TM15) along with pi-pi interaction at Tyr1160. Interestingly, other findings have also shown dihydromyricetin to be a useful and effective inhibitor for combating overexpression of MRP2 in CRC cells (Wang et al., 2021).
Our findings showed that S-(2, 4-dinitrophenyl) glutathione (-7.2 kcal/mol), MK-571(-7.1 kcal/mol), and probenecid (-6.1 kcal/mol) ranked as the third, second lowest and least with respect to binding affinity, respectively and may not be a better inhibitor among the tested drugs. S-(2, 4-dinitrophenyl) glutathione interacted with four hydrogen bonds and formed pi-alkyl and pi-sulfur interaction with the MRP2 shown in Table 3 and Fig. 3(d). MK-571 formed two hydrogen bonds, pi-alkyl interactions, and two carbon-hydrogen bonds with MRP2, as presented in Fig. 3(e) and Table 3. Our study suggested that MK-571 may not be a suitable inhibitor for overexpressed MRP2. In contrast, in vitro studies have used MK571 repeatedly to block the cellular efflux of drugs, suggesting that it acts as an inhibitor to abolish MRP2 function (Barrington et al., 2015; Munić et al., 2011; Schutte et al., 2006). Notably, probenecid showed the lowest binding affinity (-6.1 kcal/mol) with MRP2, though its interaction occurs by forming three hydrogen bonds at residues Lys495, Arg911, and Thr1090. Another in vitro study also reported that probenecid might not be considered a potent inhibitor because it failed to compete with the substrates of MRP2 (Huisman et al., 2002; Zhou et al., 2008). However, the same study suggested that probenecid may sometimes inhibit both MRP1 and MRP2, depending on substrate specificity (Huisman et al., 2002; Zhou et al., 2008). Another group has also reported that the effectiveness of probenecid as an inhibitor to perturb MRP2-mediated transport depends on the nature of the substrate that it competes with during transportation (Zelcer et al., 2003).
Interestingly, our findings showed that most of the binding sites are common in anticancer drugs and inhibitors; irinotecan binds with Thr912. This is the common binding site for Montelukast, Dihydromyricetin, and MK-571, which is part of TMD12 of MRP2 shown in Fig. 4. Similarly, Irinotecan binds at residue Arg1156, whereas dihydromyricetin also binds TM15. We also found that some anticancer drugs and inhibitors share common binding sites, as shown in Table 4. Of note, Montelukast has common binding sites, including Estradiol-17-beta-glucuronide, SN-38, irinotecan, Doxorubicin, Capecitabine, Oxaliplatin, and Gemcitabine, which suggests that Montelukast may be administered as an inhibitor, prior to the mentioned drugs, in a combinatorial approach to reduce transport or efflux of drugs from cancer cells. However, none of these inhibitors have been clinically recommended for use in combination with anticancer drugs because they lack sufficient efficacy, impermissible toxicity, and unpredictable pharmacokinetic interactions (Wang et al., 2015). The molecular docking analysis of MRP2 with anticancer drugs suggests that 5-FU may be an effective treatment for MRP2-resistant CRC. Whereas the anticancer drugs, which share common binding sites such as irinotecan/oxaliplatin and inhibitors (Montelukast and zafirlukast), could be used in a combinatorial approach and may offer an efficient strategy to combat drug resistance in CRC. Thus, implying a combinatorial approach to anticancer drugs and inhibitors/modulator interactions, can be further validated in in vitro studies to provide valuable insights into overcoming MDR in CRC by enhancing drug retention within cancer cells. Thus, our primary findings indicate that combining MRP2 inhibitors with chemotherapeutics could lead to more effective, personalized treatment strategies for resistant CRC. However, before drawing a definite conclusion further in vitro and in vivo, studies are crucial to validate the efficacy of these anticancer drugs and inhibitors to re-assess their potential as a substrate or modulator/inhibitor of MRP2.
| Common binding site | Anticancer drugs | Inhibitor |
|---|---|---|
| Gln429 | Doxorubicin, Oxaliplatin, Gemcitabine | Dihydromyricetin, MK-571, S-(2,4-dinitrophenyl) glutathione |
| Asp884 | Gemcitabine | Montelukast, Dihydromyricetin, Zafirlukast |
| Ser908 | Doxorubicin | Dihydromyricetin, MK-571, S-(2,4-dinitrophenyl) glutathione |
| Arg911 | Gemcitabine, Capecitabine | Zafirlukast |
| Ser1153 | Doxorubicin | Dihydromyricetin, MK-571, S-(2,4-dinitrophenyl) glutathione |
| Arg1156 | Irinotecan, Oxaliplatin | Montelukast, MK-571, S-(2,4-dinitrophenyl) glutathione |
| Ser1164 | Irinotecan | Montelukast, MK-571, S-(2,4-dinitrophenyl) glutathione |
Our molecular docking study provides a primary platform to shorten drug development time and increase the success rate of existing approved drugs in clinical trials. It also emphasizes the need to carry out future in silico and in vitro studies to predict and validate drug binding sites of MRP2 protein with marketed drugs as potential anticancer drug. Hence, it would be an appropriate approach for improving the efficacy of chemotherapeutic drugs to treat cancer patients.
5. Conclusions
Overexpressed MRP2 is a major detrimental factor in the efficacy of anticancer drugs, as it effluxes the drugs causing MDR in CRC cells. Our molecular docking analysis showed that irinotecan possessed the highest binding affinity, and 5-FU had the lowest binding affinity with MRP2. Irinotecan was a potential substrate due to its strong binding affinity with MRP2. Thus, we conclude that Irinotecan could not be a preferable chemotherapeutic option to treat MRP2 overexpressed CRC. In contrast, our findings revealed that 5-FU interacts weakly with MRP2, suggesting it could be a promising chemotherapeutic drug to manage resistant CRC in patients due to MRP2 over-expression. Additionally, our interaction analysis using inhibitors showed montelukast and zafirlukast to demonstrate strong binding affinity to MRP2, hence implying that a combinatorial approach to MDR cancer with irinotecan/oxaliplatin and these inhibitors could be a better strategy to overcome MRP2-related drug resistance as they share common binding sites in TM12 and TM15. It is apparent that in-silico studies have certain limitations in terms of predicting interaction, such as the inability to fully capture dynamic conformational changes and the dependence on simplified scoring functions. It necessitates further validation of such protein-ligand interactions in physiological conditions. Thus, in vitro studies (especially an efflux assay) will be indispensable in validating the anticancer drug and inhibitor interaction with MRP2 to assess the efficacy of these together used as a combinatorial approach to overcome drug resistance in MRP2 overexpressed CRC. This could lead to the development of more efficient treatment approaches for overcoming MDR in CRC patients, ultimately improving their prognosis.
CRediT authorship contribution statement
Absarul Haque: Conceptualization, Writing-Original draft preparation, Writing-Reviewing and Editing, Supervision. Ghazanfar Ali Baig: Writing-Original draft preparation, Methodology, Software. Abdulelah Saleh Alshawli: Data curation, Methodology. Mohammad Alharthi: Investigation, Writing-Reviewing and Editing. Imran Naseer: Formal analysis, Writing-Reviewing and Editing. Peter Natesan Pushparaj: Methodology, Software. Mahmood Rasool: Writing- Reviewing and Editing, Validation. F A Dain Md Opo: visualization, formal analysis.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Declaration of Generative AI and AI-assisted technologies in the writing process
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Funding
This research was funded by Institutional Fund Projects under grant no. (IFPRC-159-290-2020). The authors gratefully acknowledge the technical and financial support provided by the Ministry of Education and King Abdulaziz University, Jeddah, Saudi Arabia.
References
- The clinical relevance of gene expression based prognostic signatures in colorectal cancer. Biochim. Biophys. Acta Rev. Cancer. 2021;1875:188513. https://doi.org/10.1016/j.bbcan.2021.188513
- [CrossRef] [PubMed] [Google Scholar]
- High ABCC2 and low ABCG2 gene expression are early events in the colorectal adenoma-carcinoma sequence. PLoS One. 2015;10:e0119255. https://doi.org/10.1371/journal.pone.0119255
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Drug resistance-associated markers p-glycoprotein, multidrug resistance-associated protein 1, multidrug resistance-associated protein 2, and lung resistance protein as prognostic factors in ovarian carcinoma. Clin. Cancer Res.. 1999;5:2798-805.
- [PubMed] [Google Scholar]
- Multi drug resistance in colorectal cancer- approaches to overcome, advancements and future success. Adv. Cancer Biol. Metastasis. 2024;10 https://doi.org/10.1016/j.adcanc.2024.100114
- [CrossRef] [Google Scholar]
- A phase 2 trial of gemcitabine and docetaxel in patients with metastatic colorectal adenocarcinoma with methylated checkpoint with forkhead and ring finger domain promoter and/or microsatellite instability phenotype. Clin. Transl. Sci.. 2021;14:954-63. https://doi.org/10.1111/cts.12960
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- MK571 inhibits phase-2 conjugation of flavonols by caco-2/TC7 cells, but does not specifically inhibit their apical efflux. Biochem. Pharmacol.. 2015;95:193-200. https://doi.org/10.1016/j.bcp.2015.03.005
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The protein data bank. Nucleic Acids Res.. 2000;28:235-42. https://doi.org/10.1093/nar/28.1.235
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Transport-Mediated oxaliplatin resistance associated with endogenous overexpression of MRP2 in caco-2 and PANC-1 cells. Cancers (Basel). 2019;11:1330. https://doi.org/10.3390/cancers11091330
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Mammalian ABC transporters in health and disease. Annu. Rev. Biochem.. 2002;71:537-92. https://doi.org/10.1146/annurev.biochem.71.102301.093055
- [CrossRef] [PubMed] [Google Scholar]
- Trifluridine/Tipiracil: A review in metastatic colorectal cancer. Drugs. 2016;76:1393-402. https://doi.org/10.1007/s40265-016-0633-9
- [CrossRef] [PubMed] [Google Scholar]
- Molecular markers in colorectal cancer: Genetic bases for a customised treatment. Clin. Transl. Oncol.. 2007;9:549-54. https://doi.org/10.1007/s12094-007-0102-8
- [CrossRef] [PubMed] [Google Scholar]
- Genomic evolution and diverse models of systemic metastases in colorectal cancer. Gut. 2022;71:322-32. https://doi.org/10.1136/gutjnl-2020-323703
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Effect of multidrug resistance-reversing agents on transporting activity of human canalicular multispecific organic anion transporter. Mol. Pharmacol.. 1999;56:1219-28. https://doi.org/10.1124/mol.56.6.1219
- [CrossRef] [PubMed] [Google Scholar]
- 5-FU promotes stemness of colorectal cancer via p53-mediated WNT/β-catenin pathway activation. Nat. Commun.. 2020;11:5321. https://doi.org/10.1038/s41467-020-19173-2
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Multispecific organic anion transporter is responsible for the biliary excretion of the camptothecin deriva-tive irinotecan and its metabolites in rats. J. Pharmacol. Exp. Ther.. 1997;281:304-14.
- [PubMed] [Google Scholar]
- Role of oxaliplatin in the treatment of colorectal cancer. Ther. Clin. Risk Manag.. 2009;5:229-38. https://doi.org/10.2147/tcrm.s3583
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Drug resistance and ATP-dependent conjugate transport mediated by the apical multidrug resistance protein, MRP2, permanently expressed in human and canine cells. Mol. Pharmacol.. 1999;55:929-37.
- [PubMed] [Google Scholar]
- Small-molecule library screening by docking with pyRx. Methods Mol. Biol.. 2015;1263:243-50. https://doi.org/10.1007/978-1-4939-2269-7_19
- [CrossRef] [PubMed] [Google Scholar]
- Binding site interactions of modulators of breast cancer resistance protein, multidrug re-sistance-associated protein 2, and P-glycoprotein activity. Mol. Pharm.. 2020;17:2398-410. https://doi.org/10.1021/acs.molpharmaceut.0c00155
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Estradiol 3-glucuronide is transported by the multidrug re-sistance-associated protein 2 but does not activate the allosteric site bound by estradiol 17-glucuronide. Drug Metab. Dispos.. 2004;32:1139-45. https://doi.org/10.1124/dmd.104.000372
- [CrossRef] [PubMed] [Google Scholar]
- The MRP-related and BCRP/ABCG2 multidrug resistance proteins: Biology, substrate specificity and regulation. Curr. Drug Metab.. 2004;5:21-53. https://doi.org/10.2174/1389200043489199
- [CrossRef] [PubMed] [Google Scholar]
- Multidrug resistance protein 2-mediated estradi-ol-17βcuronide transport potentiation: In vitro-in vivo correlation and species specificity. Drug Metab. Dispos.. 2009;37:794-801. https://doi.org/10.1124/dmd.108.023895
- [CrossRef] [PubMed] [Google Scholar]
- Characterization of 5(6)-carboxy-2,′7′-dichlorofluorescein transport by MRP2 and utilization of this substrate as a fluorescent surrogate for LTC4. J. Biomol. Screen. 2008;13:295-301. https://doi.org/10.1177/1087057108316702
- [CrossRef] [PubMed] [Google Scholar]
- MRP2 (ABCC2) transports taxanes and confers paclitaxel resistance and both processes are stim-ulated by probenecid. Int. J. Cancer. 2005;116:824-9. https://doi.org/10.1002/ijc.21013
- [CrossRef] [PubMed] [Google Scholar]
- MRP2 (ABCC2) transports taxanes and confers paclitaxel resistance and both processes are stimulated by probenecid. Int. J. Cancer. 2005;116:824-9. https://doi.org/10.1002/ijc.21013
- [CrossRef] [PubMed] [Google Scholar]
- Multidrug resistance protein 2 (MRP2) transports HIV protease inhib-itors, and transport can be enhanced by other drugs. AIDS. 2002;16:2295-301. https://doi.org/10.1097/00002030-200211220-00009
- [CrossRef] [PubMed] [Google Scholar]
- Mutation of Trp1254 in the multispecific organic anion transporter, multidrug resistance protein 2 (MRP2) (ABCC2), alters substrate specificity and results in loss of methotrexate transport activity. J. Biol. Chem.. 2001;276:38108-14. https://doi.org/10.1074/jbc.M105160200
- [CrossRef] [PubMed] [Google Scholar]
- Efflux transport of estrogen glucuronides by human MRP2, MRP3, MRP4 and BCRP. J. Steroid Biochem. Mol. Biol.. 2018;178:99-107. https://doi.org/10.1016/j.jsbmb.2017.11.007
- [CrossRef] [PubMed] [Google Scholar]
- Bile acid metabolism during development. In: Fetal and Neonatal Physiology. Vol Volume 2. Elsevier; 2017. p. :913-929.e4. https://doi.org/10.1016/B978-0-323-35214-7.00095-0
- [Google Scholar]
- The roles of MRP2, MRP3, OATP1B1, and OATP1B3 in conjugated hyper-bilirubinemia. Drug Metab. Dispos.. 2014;42:561-5. https://doi.org/10.1124/dmd.113.055772
- [CrossRef] [PubMed] [Google Scholar]
- Didox and resveratrol sensitize colorectal cancer cells to doxorubicin via activating apoptosis and ameliorating p-glycoprotein activity. Sci. Rep.. 2016;6:36855. https://doi.org/10.1038/srep36855
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Double-edged sword of chemosensitizer: Increase of multidrug resistance protein (MRP) in leukemic cells by an MRP inhibitor probenecid. Biochem. Biophys. Res. Commun.. 2001;283:64-71. https://doi.org/10.1006/bbrc.2001.4746
- [CrossRef] [PubMed] [Google Scholar]
- PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res.. 2019;47:D1102-9. https://doi.org/10.1093/nar/gky1033
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Multidrug resistance-associated protein 2 determines the efficacy of cisplatin in patients with hepatocellular carcinoma. Oncol. Rep.. 2010;23:965-72. https://doi.org/10.3892/or_00000721
- [CrossRef] [PubMed] [Google Scholar]
- Improving protein-ligand binding site prediction accuracy by clas-sification of inner pocket points using local features. J. Cheminform. 2015;7 https://doi.org/10.1186/s13321-015-0059-5
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The MRP family of drug efflux pumps. Oncogene. 2003;22:7537-52. https://doi.org/10.1038/sj.onc.1206953
- [CrossRef] [PubMed] [Google Scholar]
- Overexpression of cMOAT (MRP2/ABCC2) is associated with decreased formation of platinum-DNA adducts and decreased G2-arrest in melanoma cells resistant to cisplatin. J. Invest. Dermatol.. 2003;121:172-6. https://doi.org/10.1046/j.1523-1747.2003.12313.x
- [CrossRef] [PubMed] [Google Scholar]
- ATP-dependent 17β-Estradiol 17-(β-D-Glucuronide) transport by multidrug resistance protein (MRP): Inhibition By Cholestatic Steroids. J. Biol. Chem.. 1996;271:9683-9689. https://doi.org/10.1074/JBC.271.16.9683
- [CrossRef] [PubMed] [Google Scholar]
- Location of the rhodamine-binding site in the human multidrug resistance p-glycoprotein. J. Biol. Chem.. 2002;277:44332-8. https://doi.org/10.1074/jbc.M208433200
- [CrossRef] [PubMed] [Google Scholar]
- Drug resistance in colorectal cancer: An epigenetic overview. Biochim. Biophys. Acta Rev. Cancer. 2021;1876:188623. https://doi.org/10.1016/j.bbcan.2021.188623
- [CrossRef] [PubMed] [Google Scholar]
- Inhibition of multi-drug resistance of ovarian carcinoma by small interfering RNA targeting to MRP2 gene. Arch. Gynecol. Obstet.. 2009;279:149-57. https://doi.org/10.1007/s00404-008-0690-8
- [CrossRef] [PubMed] [Google Scholar]
- Liver and gastrointestinal cancers. In: Sosnik A., Bendayan R., eds. Cancer Sensitizing Agents for Chemotherapy; Drug efflux pumps in cancer resistance pathways: From molecular recognition and characterization to possible inhibition strategies in chemotherapy. Vol Volume 7. Academic Press; 2020. p. :197-250. https://doi.org/10.1016/B978-0-12-816434-1.00007-3
- [Google Scholar]
- Intratumor heterogeneity: The rosetta stone of therapy resistance. Cancer Cell. 2020;37:471-84. https://doi.org/10.1016/j.ccell.2020.03.007
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Protection of platinum-DNA adduct for-mation and reversal of cisplatin resistance by anti-MRP2 hammerhead ribozymes in human cancer cells. Int. J. Cancer. 2005;115:393-402. https://doi.org/10.1002/ijc.20899
- [CrossRef] [PubMed] [Google Scholar]
- RNA interference-triggered reversal of ABCC2-dependent cisplatin resistance in human cancer cells. Biochem. Biophys. Res. Commun.. 2006;348:153-7. https://doi.org/10.1016/j.bbrc.2006.07.022
- [CrossRef] [PubMed] [Google Scholar]
- Trifluridine/tipiracil overcomes the resistance of human gastric 5-fluorouracil-refractory cells with high thymidylate synthase expression. Oncotarget. 2018;9:13438-50. https://doi.org/10.18632/oncotarget.24412
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Concise update on colorectal cancer epidemi-ology. Ann. Transl. Med.. 2019;7:609. https://doi.org/10.21037/atm.2019.07.91
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Structural basis for the modulation of MRP2 activity by phos-phorylation and drugs. Nat. Commun.. 2024;15:1983. https://doi.org/10.1038/s41467-024-46392-8
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Lack of association between expression of MRP2 and early relapse of colorectal cancer in patients receiving FOLFOX-4 chemotherapy. Oncol. Lett.. 2012;4:893-7. https://doi.org/10.3892/ol.2012.889
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Molecular docking. Methods Mol. Biol.. 2008;443:365-82. https://doi.org/10.1007/978-1-59745-177-2_19
- [CrossRef] [PubMed] [Google Scholar]
- Characterization of rhodamine-123, calcein and 5(6)-carboxy-2′, 7′-dichlorofluorescein (CDCF) export via MRP2 (ABCC2) in MES-SA and A549 cells. Eur. J. Pharm. Sci.. 2011;43:359-69. https://doi.org/10.1016/j.ejps.2011.05.003
- [CrossRef] [PubMed] [Google Scholar]
- Identification of MRP2 as a targetable factor limiting oxaliplatin accumulation and response in gastrointestinal cancer. Sci. Rep.. 2019;9:2245. https://doi.org/10.1038/s41598-019-38667-8
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Multidrug resistance-associated protein 2 (MRP2) mediated transport of oxaliplatin-derived platinum in membrane vesicles. PLoS One. 2015;10:e0130727. https://doi.org/10.1371/journal.pone.0130727
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- 5-Fluorouracil-based chemotherapy for colorectal cancer and MTHFR/MTRR genotypes. Br. J. Clin. Pharmacol.. 2011;72:162-3. https://doi.org/10.1111/j.1365-2125.2010.03892.x
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The role of capecitabine in the treatment of colorectal cancer in the elderly. Anticancer Res.. 2006;26:2381-6.
- [PubMed] [Google Scholar]
- Trifluridine/Tipiracil and regorafenib in patients with metastatic colorectal cancer: A retrospective study at a tertiary oncology center. Oncologist. 2021;26:e2161-9. https://doi.org/10.1002/onco.13942
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Identification of FDA-approved drugs as modulators of multidrug resistance protein 2 (MRP2/ABCC2) expression levels in MRP2-overexpressing cells: Preliminary Data. J. Biosci. Med. (Irvine). 2024;12:32-44. https://doi.org/10.4236/jbm.2024.1210004
- [CrossRef] [Google Scholar]
- Combination chemotherapy against colorectal cancer cells: Co-delivery of capecitabine and pioglitazone hydrochloride by polycaprolactone-polyethylene glycol carriers. Life Sci.. 2023;332:122083. https://doi.org/10.1016/j.lfs.2023.122083
- [CrossRef] [PubMed] [Google Scholar]
- Montelukast is a potent and durable inhibitor of multidrug resistance protein 2-mediated efflux of taxol and saquinavir. Biol. Pharm. Bull.. 2009;32:2002-9. https://doi.org/10.1248/bpb.32.2002
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Identification of basic residues involved in drug export function of human multidrug resistance-associated protein 2. J. Biol. Chem.. 2000;275:39617-24. https://doi.org/10.1074/jbc.M005149200
- [CrossRef] [PubMed] [Google Scholar]
- An in vitro and in silico study on the flavonoid-mediated modulation of the transport of 2-amino-1-methyl-6-phenylimidazo [4, 5-b] pyridine (PhIP) through Caco-2 monolayers. Tox-icol. Appl. Pharmacol.. 2006;217:204-15. https://doi.org/10.1016/j.taap.2006.08.005
- [Google Scholar]
- Colorectal cancer statistics, 2023. CA. Cancer J. Clin.. 2023;73:233-54. https://doi.org/10.3322/caac.21772
- [CrossRef] [PubMed] [Google Scholar]
- Dubin-Johnson syndrome coinciding with colon cancer and atherosclerosis. World J. Gastroenterol.. 2013;19:946-50. https://doi.org/10.3748/wjg.v19.i6.946
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Evodiamine suppresses ABCG2 mediated drug re-sistance by inhibiting p50/p65 NF-κB pathway in colorectal cancer. J. Cell Biochem.. 2016;117:1471-81. https://doi.org/10.1002/jcb.25451
- [CrossRef] [PubMed] [Google Scholar]
- BIOVIA discovery studio dassault syst mes BIOVIA, Discovery Studio Modeling Environment.
- Identification and characterisation of putative drug binding sites in human ATP-binding cassette B5 (ABCB5) transporter. Comput. Struct. Biotechnol. J.. 2020;19:691-704. https://doi.org/10.1016/j.csbj.2020.12.042
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Cetuximab enhanced the efficacy of chemotherapeutic agent in ABCB1/P-glycoprotein-overexpressing cancer cells. Oncotarget. 2015;6:40850-65. https://doi.org/10.18632/oncotarget.5813
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- PubChem: A public information system for analyzing bioactivities of small molecules. Nucleic Acids Res.. 2009;37:W623-33. https://doi.org/10.1093/nar/gkp456
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Dihydromyricetin reverses MRP2-induced multidrug resistance by pre-venting NF-κB-Nrf2 signaling in colorectal cancer cell. Phytomedicine. 2021;82:153414. https://doi.org/10.1016/j.phymed.2020.153414
- [CrossRef] [PubMed] [Google Scholar]
- Reverting doxorubicin resistance in colon cancer by targeting a key signaling protein, steroid receptor coactivator. Exp. Ther. Med.. 2018;15:3751-8. https://doi.org/10.3892/etm.2018.5912
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Evidence for two interacting ligand binding sites in human multidrug resistance protein 2 (ATP binding cassette C2) J. Biol. Chem.. 2003;278:23538-44. https://doi.org/10.1074/jbc.M303504200
- [CrossRef] [PubMed] [Google Scholar]
- Comprehensive analysis identifies novel targets of gemcitabine to improve chemotherapy treatment strategies for colorectal cancer. Front Endocrinol. (Lausanne). 2023;14:1170526. https://doi.org/10.3389/fendo.2023.1170526
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Slug mediates MRP2 expression in non-small cell lung cancer cells. Biomolecules. 2022;12:806. https://doi.org/10.3390/biom12060806
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Substrates and inhibitors of human multidrug resistance associated proteins and the implications in drug devel-opment. Curr. Med. Chem.. 2008;15:1981-2039. https://doi.org/10.2174/092986708785132870
- [CrossRef] [PubMed] [Google Scholar]




