

Translate this page into:
Estimation of electrostatic and covalent contributions to the enthalpy of H-bond formation in H-complexes of 1,2,3-benzotriazole with proton-acceptor molecules by IR spectroscopy and DFT calculations
⁎Corresponding author. issaoui_noureddine@yahoo.fr (Noureddine ISSAOUI), omar@ksu.edu.sa (Omar M. Al-Dossary),
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Using the methods of IR-spectroscopy and quantum chemical calculations, we determined the formation of an H-bond between the 1,2,3-benzotriazole molecule and the molecules of acetone, dioxane, DMF, and DMSO. Quantum chemistry methods have been used to calculate the sums of charge changes in the atoms of the 1,2,3-benzotriazole molecule and of proton acceptor molecules (acetone, dioxane, DMF, and DMSO) due to the formation of their complexes with an H-bond (H-complexes N—H…O). The sum of charge changes was taken as a contribution of a covalent component to H-bond formation. It has been established that as the sum of charge changes in N, H, and O or H and O, forming the H-complex, increases, the enthalpy of H-complex formation (EF) decreases. On the contrary, the EO of H-complexes increases when the product of the initial charges on the H and O atoms increases. In the absence of H-complexes, the product of charges was considered the electrostatic component of H-bond formation. The criterion of the value of H-bond EF was the value of a low-frequency shift of the initial position of the maximum IR absorption band of the N—H vibration of the 1,2,3-benzotriazole molecule relative to its position in a neutral CCl4 solvent. The results of QTAIM, NCI, and RDG analyses showed that the values of energy densities in BCPs have a positive value in the complexes formed by 1,2,3-benzotriazole with acetone, dioxane, and DMFA, and such hydrogen bonds are electrostatic in nature. In the complex formed by 1,2,3-benzotriazole and DMSO, it has a negative value and a covalent character. The data obtained have allowed for two main conclusions: (i) In the formation of the H-complexes of the 1,2,3-benzotriazole molecule with proton-acceptor molecules containing oxygen atoms, the main contribution to H-bond formation is made by the electrostatic component. (ii) The contribution of the covalent component increases with increasing enthalpy of H-bond formation.
Keywords
H-bond
1,2,3 – benzotriazole
DFT
AIM
ELF
NCI
RDG

1 Introduction
W. M. Latimer and W. H. Rodebush (Latimer and Rodebush, 1920) first introduced the concept of the “hydrogen bond“. Usually, the hydrogen bond represents by the scheme D—H⋯A. The atom “D” is bound to hydrogen by a covalent chemical bond called the hydrogen bond donor, and the atom “A“ is the hydrogen bond acceptor. At present, the concepts of hydrogen (H) bonds are widely used to explain the peculiarities of protein structure and nucleic acids, the mechanism of numerous enzyme catalyzed processes, occurring in an organism (Gerlt et al., 1997; Perrin and Nielson, 1997), and other chemical reactions, involving proton transfer (Zundel et al., 2000).
As understanding the effect of H-bonds on physicochemical molecule parameters is highly relevant for molecular biology and chemistry, the characteristics of H-bonds have been studied in many theoretical and experimental works by methods of IR spectroscopy, NMR spectroscopy, X-ray, and neutron diffraction (Allerhand and Schleyer, 1963; Hobza et al., 2000; Howard et al., 2010; Paesani et al., 2009; Wang et al., 2017; Smith et al., 2019; Brauer et al., 2005; Tuttle and Graefenstein, 2004; Allen, 2002), and more recently, theoretical and quantum-chemical studies have become widespread (Nemes et al., 2018; Gilli and Gilli, 2010; Grabowski, 2011; Reed et al., 1986; Dolgonosov, 2019; Stone, 2017).
Despite numerous studies on the effect of hydrogen bonds on the physical and chemical properties of substances, the question of which factor predominates—electrostatic (Stone, 2017; Pauling, 1960) or covalent (Nemes et al., 2018; Dolgonosov, 2019; Mulliken, 1952)—remains open.
Earlier (Mulloev et al., 2021), a semi-empirical technique had been proposed to obtain information on the contribution of the electrostatic component to hydrogen bond formation. It was based on a comparison of the data from IR spectroscopy (information about the effectiveness of H-bond formation) with the results of quantum chemical calculations of the initial charges on the H atoms of the N—H bond of pyrrole, indole, and carbazole molecules forming H-complexes with an acetone molecule (a proton acceptor), i.e., in the H-complex D—H…A (N—H…O), the charge value varied on the H atom. However, the authors (Mulloev et al., 2021) did not analyze the contribution of covalent component change to H-complex formation.
Even earlier, in Egorochkin and Voronkov (2000), a linear correlation was also established between the charge on the H atom of the phenol molecule and the enthalpy of formation of the H-bond during the formation of the H-complex with proton-acceptor molecules (Pauling, 1960; Rekik et al., 2007). The presence of a correlation allowed the authors to conclude that the electrostatic interaction plays a predominant role in the formation of the H-complex. However, the possible contribution of the covalent component to the formation of the H-complex was also not discussed. In Egorochkin and Kuznetsova (2002), using the STATGRAPHICS 3.0 non-quantum chemical program, the change in the charges on the A atom of the proton acceptor molecule (covalent component) in the phenol–proton acceptor H complex was calculated and compared with the change in the efficiency of the formation of the H complex (from the literature data on IR spectra). Based on the IR shift of the OH vibration band of phenol, the authors established that the decrease in the enthalpy of formation of the H-complex is proportional to the decrease in the charge transfer in the proton acceptor molecule, and since, in their opinion, the change in the electrostatic interaction is proportional to the change in charge, the paper concludes that the main contribution to the value of the enthalpy of formation is introduced by the electrostatic interaction, i.e., the analysis of the direct covalent and electrostatic contributions was not carried out. Thus, information on the study of changes in the electrostatic and covalent components during the formation of H-complexes of the same type was absent until recently. In this work, to estimate the contributions of the electrostatic and covalent components to the formation of the H-complex {D—H…A}, an approach is proposed, the essence of which is to compare the experimental results (IR spectra) and the data of quantum chemical calculations of the charge values on the D, H, A atoms up to and after the formation of H-complexes, i.e., the change in EO from the change in the covalent and electrostatic components is analysed separately.
In this work, a simple approach is proposed to estimate the contributions of electrostatic and covalent components to the formation of the H-complex (D-H…A).
2 The essence of the approach
A methodical method consists in comparing the change in the enthalpy of H-bond formation to the values of the change in the covalent and electrostatic components in the molecular fragment {D—H…A}. The change in the enthalpy of H-bond formation correlates well with the low-frequency shift, Δv, of the A—H vibration band in IR spectra (Δv = v0-vC, v0 and vC are the frequencies of A-H vibration without and with H-complex (Mulloev et al., 2021). Thus, the Δv value can be taken as the relative value of EF.
In the structural fragment {D—H…A} on the atoms D, H and A (Σ3i) or A and H (Σ2i) the sum of charge changes on these atoms is taken as the covalent contribution to H-bond formation.
In (1), (2), qDC, qD0 – are the charges on the atom of the proton donor molecule with and without H-complex; qHC, qH0 are the charges on the hydrogen atom of the proton donor molecule with and without H-bond; qAiC, qAi0 are the charges on the atom of the proton acceptor molecule with and without H-bond; the index “i“ means the charge value on the A atom of the i-th molecule of the proton acceptor.
The value ai can be taken as the value of absolute change of electrostatic interaction between atoms Н and Аi (3)
Thus, the approach proposed makes it possible to determine both the change in both the covalent and electrostatic contributions with varying EF value and in their ratio.
In this work, the method is probed to determine the covalent and electrostatic contributions in relation to the enthalpy of H-bond formation. To this end, 1,2,3-benzotriazole was used as a proton donor molecule. The 1,2,3-benzotriazole molecule is a typical representative of the class of molecules that form intermolecular H-bonds due to the presence of the N—H group, and for it there is spectral information on the frequency shifts of the position of the absorption band maximum during the formation of H-complexes with these molecules, except for H-complexes 1,2,3-benzotriazole–dioxane (Narziev and Mulloev, 1999). Thus, the studied H-complexes were formed by the same atomic structural fragment {N—H…O}, but having a different initial charge on the O acceptor atom (Table 1).
A proton-acceptor molecule
qN0,
a.u.qNC,
a.u.qH0,
a.u.qHC,
a.u.qOi0,
a.u.qOiC,
a.u.Δv = v0-vC,
cm−1
Acetone
−0.361
−0.158
0.373
0.532
−0.267
−0.422
228
Dioxane
−0.361
−0.138
0.373
0.607
−0.336
−0.401
272
DMF
−0.361
−0.16
0.373
0.517
−0.502
−0.552
318
DMSO
−0.361
−0.231
0.373
0.542
−0.479
−0.474
390
3 Experimental and computational methods
3.1 Experimental
The IR spectra of 1,2,3-benzotriazole were obtained using a Specord-75 IR. The resulting spectra were digitized with the Get Data Graph Digitizer program. The concentration of 1,2,3-benzotriazole amounted to 0,0047 M. The concentration of proton acceptor molecules was 0,46 M. T = (24 ± 1)°C.
The acetone, dioxane, DMF, and DMSO solvents of the “Aldrich” brand were applied without further purification.
3.2 Computational details
All calculations were carried out in the Gaussian 09 W program (Frisch et al., 2010) using the Density functional theory (DFT) method. The DFT method, particularly with the B3LYP hybrid functional, has been used successfully to compute the energies and various physicochemical parameters of molecular complexes in various solutions (Chetry and Devi, 2021; Jumabaev et al., 2022). To investigate intermolecular hydrogen bonding, we used the B3LYP functional and the 6-311++G(d,p) basis set, which includes diffusion and polarization functionals (Ghalla et al 2018). In addition, the topological properties of the electron density distribution in the most stable structure were obtained using the MULTIWFN (Lu and Chen, 2012) program. VMD (Humphrey et al., 1996) software was used to perform non-covalent interaction (NCI) and reduced density gradient (RDG) analyzes and to plot the results.
4 Results and discussion
It is known (Muthu et al., 2013; Muthu and Prabhakaran, 2014) that the N—H stretching frequency is located in the region 3300–3500 cm−1. The N—H stretching frequency of 1,2,3-benzotriazole was determined at 3462 cm−1 (Fig. 1a). The experimental IR spectra of N—H stretching vibration of pure benzotriazole and its solutions in acetone and dioxane are shown in Fig. 1. In the process of solvation and association, intermolecular interaction forces affect the vibrational parameters of molecules (Rekik et al., 2007). Fig. 1 shows that when 1,2,3-benzotriazole is dissolved in acetone, the N—H stretching frequency shifts to 3234 cm−1, while it shifts to 3190 cm−1 when dissolved in dioxane..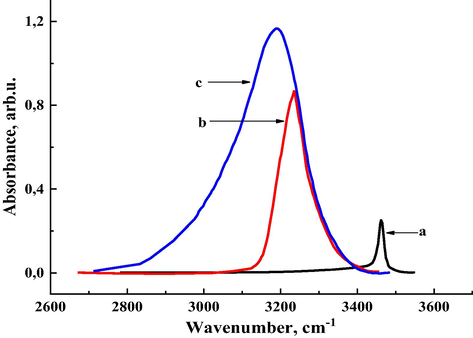
Experimental absorption IR-spectra of 1,2,3-benzotriazole monomer in the monomeric state (a) and its H-complexes with acetone (b) and dioxane (c).
In the Fig. 2, the optimal geometries and the Mulliken atomic charge distribution of 1,2,3-benzotriazole and its H-bonded complexes with acetone, dioxane, N,N-dimethylformamide (DMF), dimethylsulfoxide (DMSO) calculated at the B3LYP/6-311++G(d,p) level are described.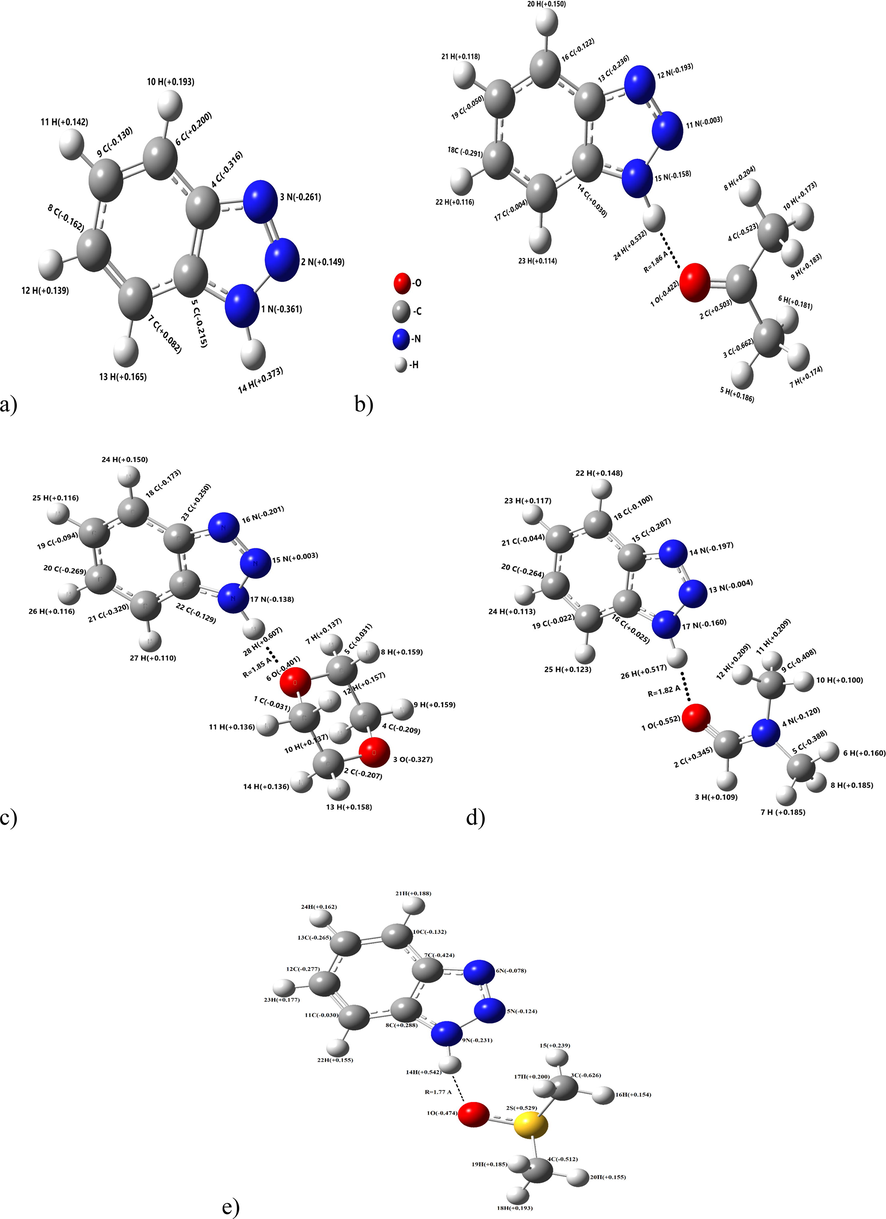
The optimal geometries and the Mulliken atomic charge distribution of 1,2,3-benzotriazole and its H-bonded complexes with acetone, dioxane, DMF, DMSO.
We can see from the Fig. 2, 1,2,3-benzotriazole molecule is formed H-bonded complexes with acetone, dioxane, DMF and DMSO molecules by N—H…O. The H-bond lengths are 1.86, 1.85, 1.82 and 1.77 Å, respectively.
Table 1 shows the Mulliken charge distribution on N and H atoms of 1,2,3-benzotriazole and O atoms of proton-acceptor molecules.
The dependences of the enthalpy of H-bond formation on the atomic charge variations on the H, N, and O atoms are demonstrated in Fig. 3. Fig. 3a plots Δv versus ΔqH. As follows from the data, there is no systematic change in Δv with ΔqH. However, it is worth noting that the sign of ΔqH is positive for all H-complexes, which indicates that the H atom is the electron donor in the formation of the H-complex.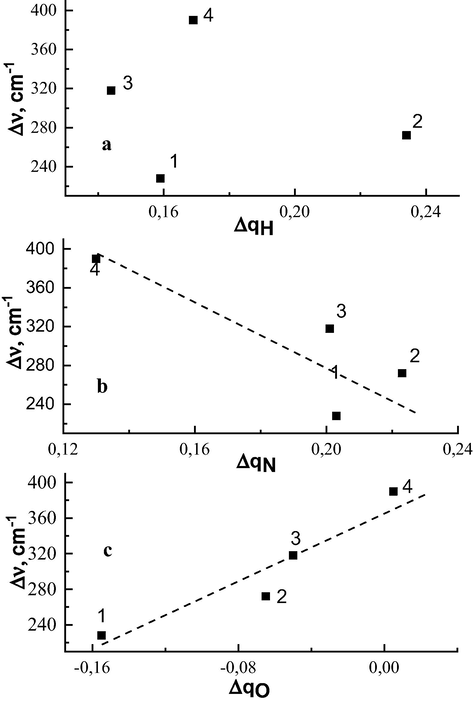
Dependence of the shift in the value of charge change on atoms H (a), N (b) of 1,2,3-benzotriazole molecule and on atom Oi (c) of proton acceptor molecules. 1 – H-complex with acetone molecule; 2 – H-complex with dioxane molecule; 3 – H-complex with DMF molecule; 4 – H-complex with DMSO molecule.
For the N atom (Fig. 3b), Δv decreases with increasing ΔqN. Thus, a negative charge decreases on the N atom with increasing enthalpy of H-bond formation in the H-complex, i.e., the N atom (as the H-atom) is also the electron donor in the formation of the H-complex.
Fig. 3c plots Δv versus ΔqOi for the Oi atoms of proton acceptor molecules. As seen from the figure, electron acceptance is recorded for the complexes of 1,2,3-benzotriazole with acetone, dioxane, and DMF (the negative charge increases), but the value of growth decreases with increasing Δv. Thus, for the H-complexes, the electron accepting ability of O atoms in a given series decreases with increasing enthalpy of H-bond formation and in the H-complex of benzotriazole with DMSO the O atom is generally the electron donor, i.e., the negative charge decreases. Note that such a decrease of the negative charge holds for the N atom of the 1,2,3-benzotriazole molecule in all H-complexes (see Table1).
Fig. 4 presents the dependences of the band shift of N—H vibration, Δv (H-bond EF), on the Σ3i values, and the insert of Fig. 4 demonstrates the dependences of the shift of the N—H vibration band, Δv (EF), on the values of Σ2i. As follows from the data, the value of Δv decreases with increasing both Σ3i and Σ2i, i.e. the enthalpy of H-bond formation decreases with increasing value of charge transfer.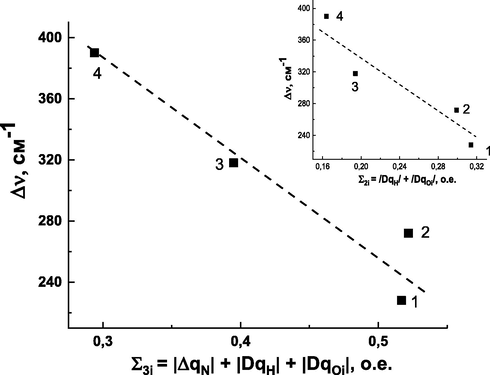
Dependence of the N—H—vibration band shift of 1,2,3-benzotriazolemolecule,Δv, on the values of Σ3i(a) and Σ2i(b) (insert). The numbers represent complexes and correspond to the designations in Fig. 3.
Fig. 5 presents the dependences of the N—H vibration band shift, Δv (the enthalpy of H-bond formation), on the product of charges on the H and O atoms. According to the data obtained, the value of the enthalpy of H-bond formation increases with increasing electrostatic component. Thus, as follows, the enthalpy of the formation of 1,2,3-benzotriazole H-complexes with proton acceptor molecules is proportional to the value of the electrostatic component. Thus, as both the covalent Σi and the electrostatic αi components increase, the EO value of the H-complexes decreases.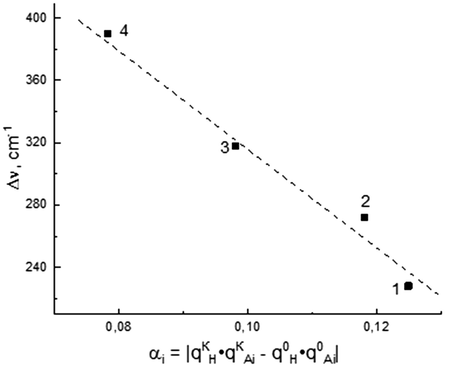
Dependence of the N—H vibration band shift of 1,2,3-benzatriazole molecule, Δv on the αi values. The numbers represent complexes and correspond to the designations in Fig. 3.
It is assumed that a broader application of the approach proposed to studying the question of the ratio between the covalent and electrostatic contributions to the formation of H-bonding to other H-complexes will favor further progress in understanding the nature of the hydrogen bond.
5 The AIM, RDG, NCI and ELF analysises
Atoms in Molecules (AIM) (Bader, 1985) theory is a very useful tool for analyzing hydrogen bonding. The formation of hydrogen bonds is associated with the presence of a critical point (BCP) between the hydrogen atom of the donor group and the acceptor atom. Poplier (Poplier and Badr, 1992) proposed a set of criteria for the presence of hydrogen bonding within the AIM formalism. The electron density ρ(r) and electron density Laplacian at the critical points of the hydrogen bond are 0.002–0.035 and 0.024–0.139 a.u., respectively (Chetry and Devi, 2021; Tang et al., 2006).
Table 2. lists some topological parameters calculated for the complexes formed by 1,2,3-benzotriazole with acetone, dioxane, DMF and DMSO molecules. It can be seen from the table that the values of electron density and Laplacian of electron density for the listed complexes are in the range of 0.0267–0.0427 au and 0.0875–0.1354 au, respectively. These values are in the range of hydrogen bonds. This means that the 1,2,3-benzotriazole molecule forms a complex with acetone, acetonitrile, dioxane, DMF and DMSO molecules by hydrogen bonding. Hydrogen bond energies were calculated using the following formula (Espinosa et al., 1998):
Complex
BCP
Electron density, ρ
Laplacian of electron density, ∇2ρ
Energy density, H
Potential energy density, V
H-bond energy, EHB
1.2.3-benzotriazole + acetone
N15-H24…O1
0.0326
0.1122
0.0008
−0.0265
8.31
1.2.3-benzotriazole + dioxane
N17-H28…O6
0.0334
0.1144
0.0003
−0.0279
8.75
1.2.3-benzotriazole + DMFA
N17-H26…O1
0.0348
0.1275
0.0006
−0.0305
9.57
1.2.3-benzotriazole + DMSO
N15-H24…O1
0.0427
0.1354
−0.0029
−0.0398
12.49
The values of energy densities in BCPs have a positive value in the complexes formed by 1,2,3-benzotriazole with acetone, dioxane and DMF, and such hydrogen bonds are electrostatic in nature. In the complex formed by 1,2,3-benzotriazole with DMSO, it takes a negative value and has a covalent character.
Fig. 6 shows the of AIM molecular graphs of the complexes of 1.2.3-benzotriazole with acetone, dioxane, DMF and DMSO. The lines linking the nuclei are the bond paths, the orange dots on the bond paths are the BCPs, and the yellow dost are the ring critical points (RCPs).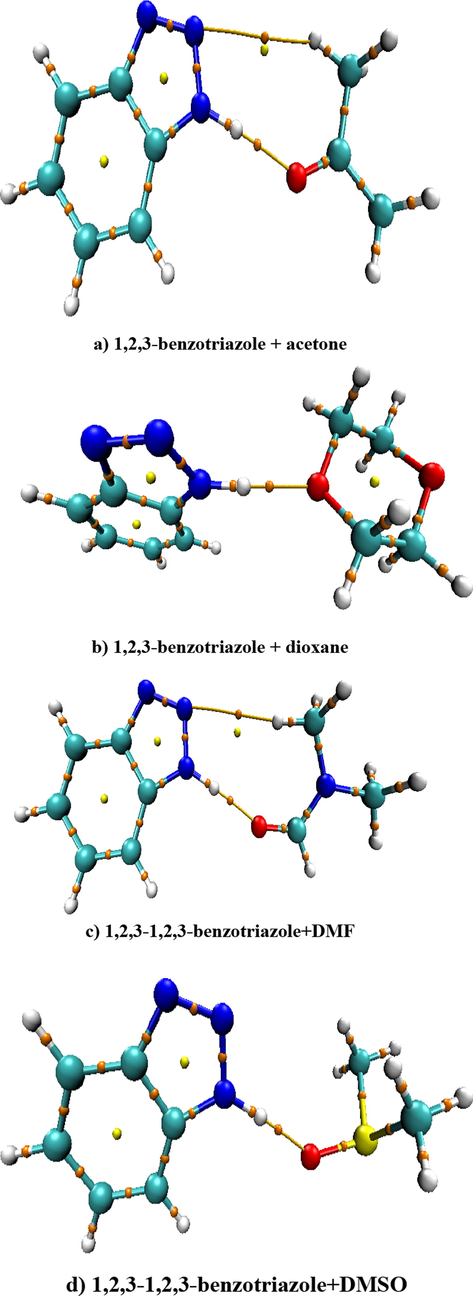
AIM molecular graphs of the complexes of 1.2.3-benzotriazole with acetone (a), dioxane (b), DMF (c), and DMSO (d).
Fig. 7 shows the results of NCI and RDG analysis of weak interactions between 1,2,3-benzotriazole and acetone, dioxane, DMF, DMSO molecules.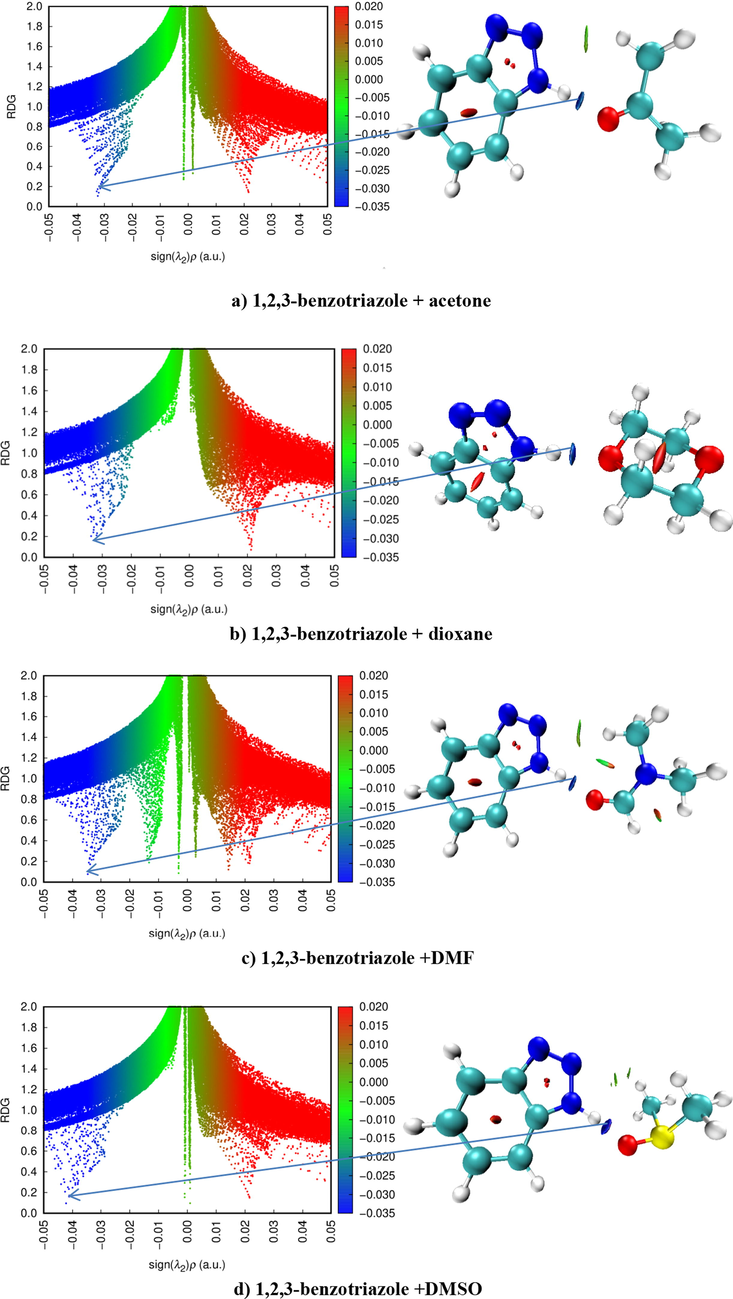
The results of NCI and RDG analyses on the 1,2,4-benzotriazole with acetone (a), dioxane (b), DMF (c) and DMSO (d).
Blue colors represent hydrogen bonding, green colors represent van der Waals interactions, and red colors represent strong repulsion (steric effect). The results of the analysis showed that there are N—H…O hydrogen bonds, C—H…N Van der Waals interactions in the complexes. Hydrogen bonding also plays a dominant role in the formation of complexes.
The electron localization function (ELF) is widely used to investigate charge transfer, classify chemical bonds on the molecular surface with a high electron path, and describe lone pairs, nuclear regions, and valence shells in atoms (Silvi et al., 2005; Fuentealba et al., 2007; Kazachenko et al., 2022). The ELF value is typically displayed in a range of colours ranging from 0.0 to 1.0. Fig. 8 represents the ELF diagrams of complexes formed by 1,2,3-benzotriazole with proton acceptor molecules (acetone, dioxane, DMF and DMSO). Charge delocalization regions (blue) with ELF < 0.5 are shown in this figure, indicating that the regions contain both bonded and non-bonded localized electrons. Electron localization sites (ELF > 0.5) describe regions where covalent electron delocalization is expected (Silvi et al., 2005).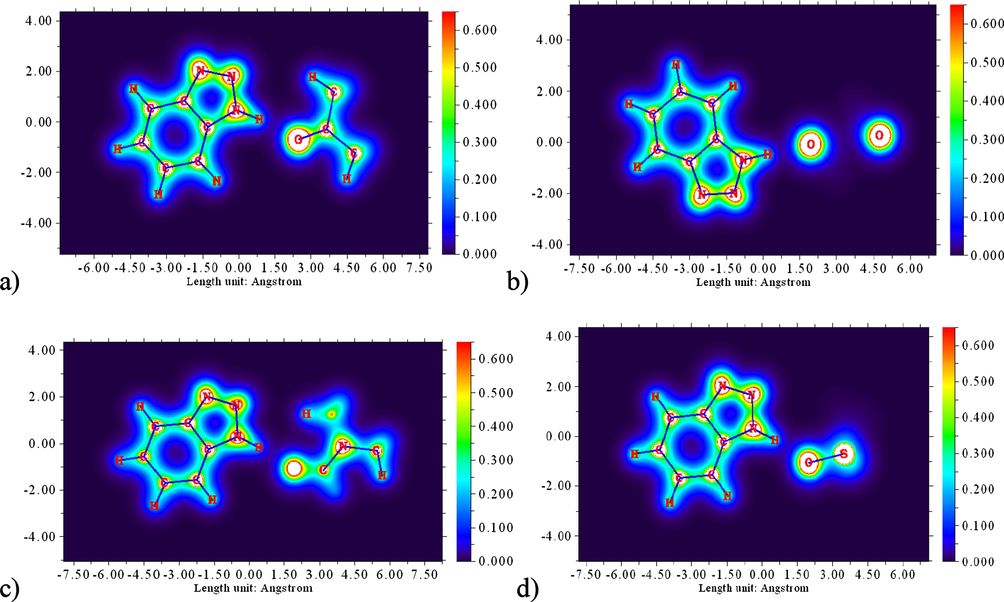
ELF diagrams of complexes of 1,2,3- benzotriazole with acetone (a), dioxane (b), DMF (c) and DMSO (d).
6 Conclusion
The results obtained using IR spectroscopy and quantum chemical calculations made it possible to conclude that the predominant contribution to the formation of the H-bond between the 1,2,3-benzotriazole molecule and the molecules of acetone, dioxane, DMF, and DMSO is made by the electrostatic component. The DFT approximation at the B3LYP/6–311++G(d,p) level was used to study the mechanism of the formation of H-complexes of the 1,2,3-benzotriazole molecule with proton-withdrawing solvents. In addition, AIM, NCI and RDG analyzes confirmed that intermolecular hydrogen bonds play a key role in the formation of H-complexes of 1,2,3-benzotriazole with various proton acceptor solvents. The values of energy densities in BCPs have a positive value in the complexes formed by 1,2,3-benzotriazole with acetone, dioxane and DMF, and such hydrogen bonds are electrostatic in nature. In the complex formed by 1,2,3-benzotriazole with DMSO, it takes a negative value and has a covalent character.
The proposed method for investigating the relationship between covalent and electrostatic contributions during H-bond formation in D—H…A H-complexes appears to contribute to a better understanding of the nature of hydrogen bond formation.
Acknowledgements
Researchers Supporting Project number (RSP2023R61), King Saud University, Riyadh, Saudi Arabia.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- The Cambridge structural database: a quarter of a million crystal structures and rising. Acta Crystallogr.B. 2002;58:380-388.
- [CrossRef] [Google Scholar]
- A survey of C-H Groups as proton donors in hydrogen bonding. J. Am. Chem. Soc.. 1963;85:1715-1723.
- [CrossRef] [Google Scholar]
- Vibrational Spectroscopy of the G· · ·C Base Pair: Experiment, Harmonic and Anharmonic Calculations, and the Nature of the Anharmonic Couplings. J. Phys. Chem. A.. 2005;109:6974-6984.
- [CrossRef] [Google Scholar]
- Intermolecular interaction study of L-Threonine in polar aprotic Solvent: Experimental and theoretical study. J. Mol. Liq.. 2021;338:116689
- [CrossRef] [Google Scholar]
- Understanding hydrogen bonding in terms of the theory of generalized charges. J. Struct. Chem.. 2019;60(11):1765-1774. (Russ)
- [CrossRef] [Google Scholar]
- Infrared spectroscopy of hydrogen bond and substituent effects in electron donor molecules. Izvestiya Acad. Sci. Chem Series. 2002;6:881.
- [CrossRef] [Google Scholar]
- Electronic structure of organic compounds of silicon, germanium, and tin. Nauka, Novosibirsk 2000:615.
- [Google Scholar]
- Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett.. 1998;285:170-173.
- [CrossRef] [Google Scholar]
- Gaussian 09. Wallingford CT: Gaussian, Inc.; 2010.
- Understanding and using the electron localization function. Comput. Theor. Chem.. 2007;19:57-85.
- [CrossRef] [Google Scholar]
- Understanding enzymic catalysis: the importance of short, strong hydrogen bonds. Chem. Biol.. 1997;4:259-267.
- [CrossRef] [Google Scholar]
- Intermolecular interactions and molecular docking investigations on 4-methoxybenzaldehyde. Computational Materials Science Volume. 2018;149:291-300.
- [CrossRef] [Google Scholar]
- Hydrogen bond models and theories: The dual hydrogen bond model and its consequences. J. Mol. Struct.. 2010;972:2-10.
- [CrossRef] [Google Scholar]
- What is the Covalency of Hydrogen Bonding? Chem. Rev.. 2011;111:2597-2625.
- [CrossRef] [Google Scholar]
- Chem. Rev.. 2000;100:4253-4264.
- [CrossRef]
- Effects of hydrogen bonding on vibrational normal modes of pyrimidine. J. Phys. Chem. A.. 2010;114:6803-6810.
- [CrossRef] [Google Scholar]
- Interaction of valine with water molecules: Raman and DFT study. Ukr. J. Phys.. 2022;67(8):602-610.
- [CrossRef] [Google Scholar]
- Hydrogen bonds interactions in biuret-water clusters: FTIR, X-ray diffraction, AIM, DFT, RDG, ELF, NLO analysis. J. King Saud Uni. Sci.. 2022;34:8.
- [CrossRef] [Google Scholar]
- Polarity and Ionization from the Standpoint of the Lewis Theory of Valence. J. Am. Chem. Soc.. 1920;42:1419-1433.
- [CrossRef] [Google Scholar]
- Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem.. 2012;33(5):580-592.
- [CrossRef] [Google Scholar]
- Molecular compounds and their spectra. II. J. Am. Chem. Soc.. 1952;74:811-824.
- [CrossRef] [Google Scholar]
- Studying the nature of hydrogen bonds of h-complexes of pyrrole derivatives with acetone according to ir spectroscopy data and quantum chemical calculations. J. Struc. Chem.. 2021;62(5):729-733.
- [CrossRef] [Google Scholar]
- Quantum mechanical, spectroscopic studies (FT-IR, FT-Raman, NMR, UV) and normal coordinates analysis on 3-([2-(diaminomethyleneamino) thiazol-4-yl] methylthio)-N′-sulfamoylpropanimidamide. Spectrochim. Acta A Mol.. 2013;108:307-318.
- [CrossRef] [Google Scholar]
- Spectrochim. Acta A Mol.. 2014;129:184-192.
- [CrossRef]
- J. Struc. Chem.. 1999;40(3):525.
- Hydrogen bonding from a valence bond theory perspective: the role of covalency. Phys. Chem. Chem. Phys.. 2018;20:20963-20969.
- [CrossRef] [Google Scholar]
- Infrared spectroscopy and hydrogen-bond dynamics of liquid water from centroid molecular dynamics with an Ab initio-based force field. J. Phys. Chem. B.. 2009;113:13118-13130.
- [CrossRef] [Google Scholar]
- The Nature of the Chemical Bond (3rd ed.). Ithaca, NY: Cornell University Press; 1960.
- “Strong” hydrogen bonds in chemistry and biology. Annu. Rev. Phys. Chem.. 1997;48:511-544.
- [CrossRef] [Google Scholar]
- The existence of an intramolecular C-H∙∙∙O hydrogen bond in creatine and carbamoyl sarcosine. Chem. Phys. Lett.. 1992;189:542-548.
- [CrossRef] [Google Scholar]
- Natural bond orbital analysis of molecular interactions: theoretical studies of binary complexes of HF, H2O, NH3, N2, O2, F2, CO and CO2 with HF, H2O and NH3. J. Chem. Phys.. 1986;84:5687-5705.
- [CrossRef] [Google Scholar]
- Infrared spectral density of hydrogen bonds within the strong anharmonic coupling theory: Quadratic dependence of the angular frequency and the equilibrium position of the fast mode. J. Mol. Struct. (Thoechem). 2007;821(1–3):58-70.
- [CrossRef] [Google Scholar]
- The topological analysis of the electron localization function. A key for a position space representation of chemical bonds. Monatshefte für Chemie. 2005;136:855-879.
- [CrossRef] [Google Scholar]
- Cis/Trans Energetics in Epoxide, Thiirane, Aziridine and Phosphirane Containing Cyclopentanols: Effects of Intramolecular O-H⋯O, S, N and P Contacts. Molecules. 2019;24
- [CrossRef] [Google Scholar]
- Natural bond orbitals and the nature of the hydrogen bond. J. Phys. Chem. A.. 2017;121:1531-1534.
- [CrossRef] [Google Scholar]
- Hydrogen bonds: relation between lengths and electron densities at bond critical points. Eur. Phys. J.. 2006;D 37:217-222.
- [CrossRef] [Google Scholar]
- A. Wu, E. Kraka, D. Cremer, Analysis of the NMR Spin–Spin coupling mechanism Across a H–Bond: nature of the H-Bond in proteins. J. Phys. Chem. B.. 2004;108:1115-1129.
- [CrossRef] [Google Scholar]
- On the nature of the halogen bond. J. Chem. Theory Comput.. 2017;10:3726-3737.
- [CrossRef] [Google Scholar]
- Advances in Chemical Physics. 2000;vol. 1:1-27.
- [CrossRef]
Further Reading
- Experimental FTIR and FT-Raman and theoretical studies on the molecular structures of monomer and dimer of 3-thiopheneacrylic acid. J. Mol. Struct.. 2017;1135:209-221.
- [Google Scholar]




