

Translate this page into:
DFT and molecular docking study of chloroquine derivatives as antiviral to coronavirus COVID-19
⁎Corresponding authors. issaoui_noureddine@yahoo.fr (Noureddine Issaoui), omar@ksu.edu.sa (Omar Al-Dossary)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
The recently emerged COVID-19 virus caused hundreds of thousands of deaths and instigated a widespread fear, threatening the world’s most advanced health security. In 2020, chloroquine derivatives are among the drugs tested against the coronavirus pandemic and showed an apparent efficacy. In the present work, the chloroquine and the chloroquine phosphate molecules have been proposed as potential antiviral for the treatment of COVID-19 diseases combining DFT and molecular docking calculations. Molecular geometries, electronic properties and molecular electrostatic potential were investigated using density functional theory (DFT) at the B3LYP/6-31G* method. As results, we found a good agreement between the theoretical and the experimental geometrical parameters (bond lengths and bond angles). The frontier orbitals analysis has been calculated at the same level of theory to determine the charge transfer within the molecule. In order to perform a better description of the FMOs, the density of states was determined. The molecular electrostatic potential maps were calculated to provide information on the chemical reactivity of molecule and also to describe the intermolecular interactions. All these studies help us a lot in determining the reactivity of the mentioned compounds. Finally, docking calculations were carried out to determine the pharmaceutical activities of the chloroquine derivatives against coronavirus diseases. The choice of these ligands was based on their antiviral activities.
Keywords
COVID-19
DFT
FMOs
MEP surfaces
Docking simulations

1 Introduction
In late December 2019, the coronavirus (Covid and R Team, 2019) was first reported in humans in Wuhan, China, and appeared as a rapidly spreading pandemic (Wang et al., 2020; Dong et al., 2020). About 46 million people worldwide have been infected as of 1, November 2020, and over 1 197 000 have died. It is worthy to mention that this pandemic has the same symptoms as a flue. Fatigue, fever, headache, runny nose and dry cough are the principal clinical symptoms of COVID-19. Thus far, there is no effective antiviral medication or vaccine against COVID-19 virus has been developed. Where the World Health Organization announced it as one of the most dangerous health catastrophes in human history (Bheenaveni, 2020) since this virus is accelerating very quickly more than predicted by experts (Al Shamsi et al., 2019). Therefore, searching for effective antiviral agents to battle against this virus is urgently needed. In this context, our investigations are destined for the development of therapeutic agents for COVID-19 diseases. Many scientists are working on the designing of efficacious antiviral agents with few aspect effects. Where recent research informed an inhibitor effect of the chloroquine and its derivatives on the growth of coronavirus (Gautret et al., 2020; Romano et al., 2020; Lecuit, 2020). Clinical trials have been done on Chinese patients COVID-19; have shown that the chloroquine has a great effect in terms of clinical results and viral clearance, in comparison to the control groups (Gautret et al., 2020). They have been proposed as a potential antiviral for the treatment of COVID-19 diseases based on their antiviral activities (Touret and X., 2020; Colson et al., 2020).
In this study, we evaluated the antiviral efficiency of two approved drugs which are chloroquine and chloroquine phosphate against the COVID-19 using molecular docking calculations. Docking is a technique of designing drug molecules via computer-aided by simulating the geometric of these molecules and their intermolecular forces (Noureddine et al., 2020a, 2020b). From this calculation, we can predict the different interactions between medications and targets which have an important role in the investigation of the mechanism of the effects of drugs. In this context, many nowadays papers is dedicated to searching in drug design using molecular docking studies (Jomaa et al., 2020; Sagaama et al., 2020a, 2020b; Issaoui et al., 2017). In the same frame, we can cite our previous paper (Romani et al., 2020) in which we used molecular docking analysis in the determination of the biological activity of the Niclosamide compound. As a result, the niclosamide is found to be a good inhibitor of the COVID-19 virus and can, therefore, be effective in controlling this disease.
The main contribution of this paper is to identify the potency of inhibition of chloroquine derivatives against COVID-19 virus by using a molecular docking study. To this end, we first determine the optimized structures of chloroquine and chloroquine phosphate molecules by using the density functional theory (DFT) at B3LYP/6-31G* level of theory. Utilizing optimized structures is more exact in docking calculations, which makes the program more trustworthy to be employed in structure-based drug design. Subsequently, their reactivities were foreseen at the same level of theory by using the frontier orbital studies (Brédas, 2014; Parr and Pearson, 1983). From this analysis, we can found the most reactive antiviral ligand. Moreover, molecular electrostatic potentials surfaces were carried out to investigate which are the most reactive nucleophilic and electrophilic regions of a molecule against reactive biological potentials. Docking calculations were performed using four structures of COVID-19 (PDB codes: 6 M03, 5R7Y, 5R81 and 6LU7) (http://www.rcsb.org/). Basing on the binding affinities and the different interactions that exist between amino acid residues and ligands, molecular docking results were discussed.
2 Computational details
2.1 DFT calculations
The GaussView program (GaussView, Guassian, Inc.) was utilized to model the initial structures of the chloroquine and the chloroquine phosphate molecules. Subsequently, their molecular geometries optimizations were carried out in the gas phase with the density functional theory (DFT) with the Gaussian 09 software package (Gaussian 09, Revision C.01, Frisch et al., 2009). All the quantum-chemical calculations have been performed via the hybrid B3LYP (Becke’s three parameter hybrid functional with Lee-Yang-Parr correlation functional LYP (Lee et al., 1988; Becke, 1993) at 6-31G* basis set. Furthermore, several electronic properties for instance the frontier molecular orbitals, gap energies, reactivity descriptors were computed using TD-DFT approach (Liu et al., 2015; Becke, 1993). The density of states (DOS) plots was obtained by using Gauss-Sum software (O'Boyle et al., 2008).
2.2 Ligands and proteins preparation
The 3D structures of COVID-19 protein were retrieved from the RCSB PDB database (http://www.rcsb.org) (http://www.rcsb.org/). The Protein Data Bank (PDB) archive contains thousand protein structures obtained either by crystallography X-ray or by NMR. Concerning ligands, the 2D structures of chloroquine and chloroquine phosphate were extracted from the PubChem online database (https://pubchem.ncbi.nlm.nih.gov/). The ligands were saved in the MDL Mol file format. Then, they were converted to a PDB file format by using Accelrys Discovery Studio Visualizer (Visualizer, 2005). Thereafter, Rapid-Screening docking was carried out using iGEMDOCK program (Yang and Chen, 2004). It is a Drug Design System for docking calculations and screening by BioXGEM labs. All the trials were docked with a population size set to 800, with 80 generations and 10 solutions.
3 Results and discussion
3.1 Optimization of chloroquine and chloroquine phosphate
Optimized structures and numbering of atoms of chloroquine and chloroquine phosphate molecules are shown graphically in Figs. 1 and 2, obtained at B3LYP/6-31G* method. Table 1 illustrates their geometrical parameters such as the calculated total energies, the dipole moments, the RMS and the maximum Cartesian force. The global minimum energies are found to be −1326.0352 a.u (≈ −36083 eV) and −2614.3242 a.u (≈ −71139) for chloroquine and chloroquine phosphate, respectively. The RMS Cartesian force values are equal to 2.412 0.10−6, 0.04067 in chloroquine and chloroquine phosphate. Their maximum Cartesian forces are found to be 8.593 0.10−6 and 0.1449. The dipole moment of a molecule is given in the form of a three-dimensional vector and which reflects the molecular charge distribution. Hence, it can be employed as a descriptor to describe the charge movement throughout the molecule. As a result of DFT/B3LYP/6-31G* calculations, the highest dipole moment was observed for the chloroquine phosphate (∼24.49 Debye) whereas the smallest one was observed for the chloroquine (∼6.05 Debye). Of course, the adding of other atoms in the geometry of the chloroquine has an influence on their stability. We can notice that the chloroquine compound becomes more stable when adding the phosphate groups since the global minimum energy decreases. Also, the strong increase in the dipole moment value shows that the chloroquine is harder before adding the phosphate groups. Moreover, it promotes the formation of hydrogen bonds.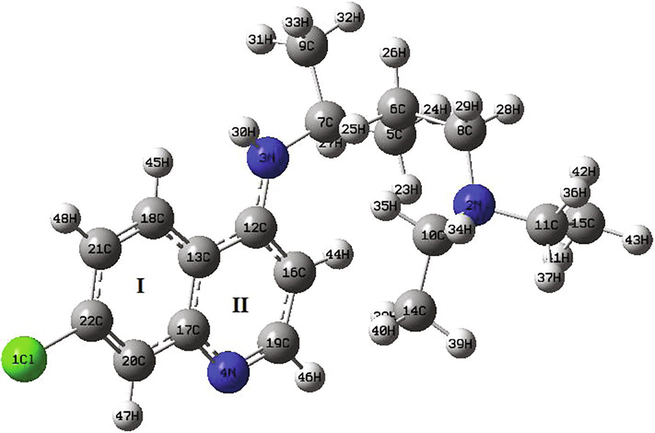
Optimized structure of the chloroquine by using DFT/B3LYP/6-31G* method.
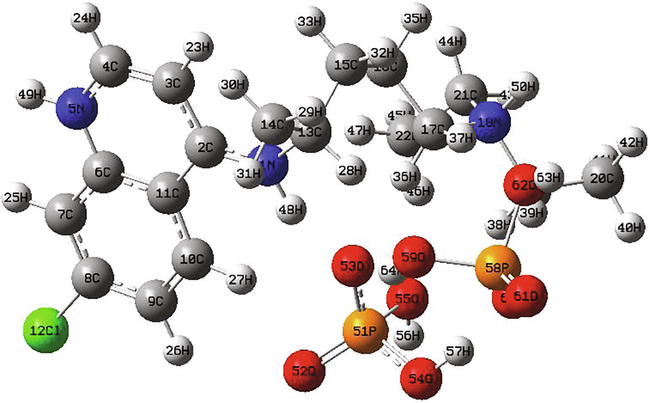
Optimized structure of the chloroquine phosphate molecule.
B3LYP/6-31G* method
Molecules
E (Hartree)
RMS Cartesian force
µ (D)
Maximum Cartesian force
Chloroquine
−1326.0352
2.412 0.10−6
6.05
8.593 0.10−6
Chloroquine phosphate
−2614.3242
0.04067
24.49
0.1449
The optimized geometrical parameters of chloroquine derivatives have been determined by the above method and they are given in Tables 2 and 3 with the experimental bond angles and bond lengths. First, we observed that the theoretical bond lengths of chloroquine compound are almost similar with the experimental results (Busetta and Courseille, 1973), since the value of RMSD is very small (0.001 Å). The same applies to the bond angles which have an RMSD value equal to 0.298°. Same thing for the chloroquine phosphate, according to the result as collected in table 3 the bond distances and bond angles show good agreement with the experimental data (Albesa-Jové et al., 2008). We find that the RMSD value is equal to 0.065 Å for the bond distances and 3.382° for the bond angles. Results reveal that the carbon–carbon bond distances are found in the range 1.374–1.546 Å for C20-C22 and C5-C7, respectively for the chloroquine. In the benzene ring (I), the carbon–carbon bond lengths C13-C17, C13-C18, C17-C20, C18-C21, C20-C22 and C21-C22 are 1.435, 1.418, 1.421, 1.378, 1.374 and 1.411 Å, respectively. The C–C bond alienation in the pyridine ring (II) is between 1.394 Å (for C12-C16 bond) and 1.445 Å (for C12-C13 bond). While, for chloroquine phosphate, the bond length between two carbon–carbon in the two rings is in the range 1.383–1.419 Å for benzene and 1.366 to 1.464 Å for pyridine ring. It is seen that the B3LYP calculated hydrogen bonding distances C–H vary from 1.009 Å (for N3-H30) to 1.099 Å (for C5-H24) for chloroquine and from 1.084 Å (for C10-H27 bond) to 1.524 Å (for C21-C22 bond) for chloroquine phosphate. Three nitrogen N atoms exist in the structure of chloroquine: the order of the N-C bond length is N2-C10 > N2-C11 > N2-C8 > N3-C7 > N3-C12 > N4-C17 > N4-C19 having values 1.470 > 1.469 > 1.467 > 1.465 > 1.370 > 1.365 > 1.319 Å, respectively. The bond distance of N3-H30 is equal to 1.009 Å. The bond angle of chloroquine between the C7-N3-H30 and C12-N3-H30 are ∼ 115.047° and ∼ 116.505°, respectively. Concerning the chloroquine phosphate, we note that the single N5-C6 bond length of 1.387 Å for ring pyridine is higher than the N5-C4 double bond (1.353 Å). The P-O bond lengths are obtained to be in range 1.489–1.693 Å (for P58-O61 and P58-O62). The O-P-O bond angles are reported in range 107.7–112.02°, whereas it is computed in range 102.543–124.278°. The C8-Cl bond length is observed at 1.743 Å and calculated at 1.748 Å. The C9-C8-Cl and C8-C9-C10 bond angles are at 119.733° and 116.940°, respectively.
Chloroquine
Parameters
Experimental
Theoretical
Parameters
Experimental
Theoretical
Bond lengths (Å)
Cl-C22
1.755
1.760
C12-C16
1.393
1.394
N2-C8
1.469
1.467
C13-C17
1.432
1.432
N2-C10
1.460
1.470
C13-C18
1.418
1.418
N2-C11
1.498
1.469
C14-H38
1.095
1.095
N3-C7
1.500
1.465
C14-H39
1.096
1.096
N3-C12
1.371
1.370
C14-H40
1.070
1.096
N3-H30
1.009
1.009
C15-H41
1.095
1.095
N4-C17
1.344
1.365
C15-H42
1.096
1.096
N4-C19
1.368
1.320
C15-H43
1.096
1.096
C5-C6
1.534
1.534
C16-C19
1.407
1.407
C5-C7
1.546
1.546
C16-H44
1.065
1.083
C5-H23
1.095
1.095
C17-C20
1.500
1.421
C5-H24
1.100
1.100
C18-C21
1.374
1.378
C6-C8
1.554
1.538
C18-H45
1.087
1.087
C6-H25
1.098
1.098
C19-H46
1.090
1.090
C6-H26
1.099
1.098
C20-C22
1.374
1.374
C7-C9
1.546
1.533
C20-H47
1.034
1.084
C7-H27
1.097
1.097
C21-C22
1.411
1.411
C8-H28
1.096
1.096
C21-H48
1.084
1.084
C8-H29
1.149
1.108
C10-H35
1.078
1.095
C9-H31
1.095
1.095
C11-C15
1.319
1.530
C9-H32
1.095
1.095
C11-H36
1.208
1.108
C9-H33
1.097
1.097
C11-H37
1.056
1.095
C10-C14
1.525
1.530
C12-C13
1.442
1.445
C10-H34
1.108
1.108
RMSD
0.001 Å
Bond angles (°)
C8-N2-C10
112.84
112.103
C15-C11-H37
108.29
108.196
C8-N2-C11
112.23
112.200
H36-C11-H37
105.89
106.039
C10-N2-C11
111.78
111.972
N3-C12-C13
120.83
120.095
C7-N3-C12
124.77
125.707
N3-C12-C16
124.34
123.092
C7-N3-H30
115.049
115.048
C13-C12-C16
116.790
116.790
C12-N3-H30
116.50
116.505
C12-C13-C17
117.68
117.797
C17-N4-C19
116.07
116.079
C12-C13-C18
124.08
123.818
C6-C5-C7
115.89
115.643
C17-C13-C18
118.16
118.383
C6-C5-H23
107.62
107.782
C10-C14-H38
110.36
110.369
C6-C5-H24
109.60
109.535
C10-C14-H39
113.36
112.214
C7-C5-H23
109.22
109.218
C10-C14-H40
110.08
110.289
C7-C5-H24
107.15
107.798
H38-C14-H39
107.9(1)
107.900
H23-C5-H24
106.4(4)
106.496
H38-C14-H40
108.5(3)
108.529
C5-C6-C8
112.5(4)
112.597
H39-14-H40
107.410
107.410
C5-C6-H25
109.59
109.519
C11-C15-H41
110.79
110.273
C5-C6-H26
110.24
110.944
C11-C15-H42
112.3(4)
112.316
C8-C6-H25
109.78
109.513
C11-C15-H43
110.2(4)
110.276
C8-C6-H26
107.46
107.750
H41-C15-H42
107.8(3)
107.894
H25-C6-H26
105.39
106.310
H41-C15-H43
108.5(4)
108.564
N3-C7-C5
113.57
113.473
H42-C15-H43
107.3(4)
107.389
N3-C7-C9
108.38
108.232
C12-C16-C19
119.7354
119.736
N3-C7-H27
106.58
106.584
C12-C16-H44
121.70
121.300
C5-C7-C9
113.83
113.289
C19-C16-H44
118.952
118.959
C5-C7-H27
107.54
107.546
N4- C17-C13
123.19
123.911
C9-C7-H27
107.33
107.331
N4- C17-C20
116.9(4)
116.950
N2-C8-C6
113.41
113.409
C13-C17-C20
119.17
119.139
N2-C8-H28
108.0(3)
108.072
C13-C18-C21
121.72
121.739
N2-C8-H29
111.43
111.344
C13-C18-H45
120.37
120.684
C6-C8-H28
108.78
108.140
C21-C18-H45
117.562
117.561
C6-C8-H29
109.59
109.436
N4-C19-C16
125.27
125.662
C28-C8-H29
106.122
106.122
N4-C19-H46
114.49
115.975
C7-C9-H31
110.742
110.742
C16-C19-H46
118.3591
118.359
C7-C9-H32
110.415
110.415
C17-C20-C22
120.35
120.214
C7-C9-H33
111.15
111.585
C17-C20-H47
117.19
117.802
H31-C9-H32
108.71
108.463
C22-C20-H47
121.70
121.984
H31-C9-H33
108.060
108.060
C18-C21-C22
119.01
119.067
H32-C9-H33
107.450
107.450
C18-C21-H48
119.39
120.983
N2-C10-C14
112.12
113.052
C22-C21-H48
119.29
119.949
N2-C10-H34
111.99
111.009
Cl-C22-C20
119.41
119.987
N2-C10-H35
107.22
107.936
Cl-C22-C21
118.84
118.570
C14-C10-H34
110.2(2)
110.284
C20-C22-C21
121.72
121.442
C14-C10-H35
109.41
108.216
N2-C11-H36
111.71
111.218
H34-C10-H35
105.45
106.030
N2-C11-H37
107.46
107.769
N2-C11-C15
113.3(2)
113.224
C15-C11-H36
110.066
110.065
RMSD
0.298°
Chloroquine phosphate
Parameters
Experimental
Theoretical
Parameters
Experimental
Theoretical
Bond lengths (Å)
N1-C2
1.409(2)
1.324
C17-N18
1.5069(6)
1.523
N1-C13
1.4967(9)
1.486
C17-H36
0.9994
1.095
N1-H48
1.0018
1.048
C17-H37
1.0005
1.094
C2-C3
1.415(3)
1.433
N18-C19
1.4980(6)
1.532
C2-C11
1.402(2)
1.464
N18-C21
1.5083(6)
1.516
C3-C4
1.400(3)
1.366
N18-H50
0.9995
1.025
C3-H23
1.000
1.079
C19-C20
1.5171(5)
1.521
C4-N5
1.366(1)
1.353
C19-H38
1.0010
1.091
C4-H24
0.999
1.084
C19-H39
1.0000
1.095
N5-C6
1.382(3)
1.387
C20-H40
1.0001
1.094
N5-H49
0.998
1.011
C20-H41
1.0001
1.096
C6-C7
1.403(1)
1.403
C20-H42
1.0000
1.098
C6-C11
1.417(3)
1.419
C21-C22
1.5296(5)
1.524
C7-C8
1.411(3)
1.386
C21-H43
1.0000
1.093
C7-H25
0.997
1.086
C21-H44
0.9998
1.094
C8-C9
1.396(3)
1.403
C22-H45
0.9998
1.095
C8-Cl
1.743(3)
1.749
C22-H46
1.0009
1.092
C9-C10
1.373(1)
1.383
C22-H47
1.0002
1.093
C9-H26
0.999
1.085
H48-O53
1.517(8)
1.675
C10-C11
1.431(3)
1.412
P51-O52
1.513(5)
1.497
C10-H27
1.001
1.084
P51-O53
1.574(5)
1.548
C13-C14
1.5142(6)
1.536
P51-O54
1.560(5)
1.594
C13-C15
1.5417(7)
1.545
P51-O55
1.000
1.682
C13-H28
0.9998
1.093
O53-H64
1.554
1.782
C14-H29
0.9993
1.095
O54-H57
0.997
1.017
C14-H30
1.0000
1.095
O55-H56
0.9969
0.972
C14-H31
1.0002
1.094
H57-O60
1.5851
1.626
C15-C16
1.5092(6)
1.544
P58-H59
1.566(6)
1.645
C15-H32
1.0002
1.097
P58-O60
1.519(5)
1.528
C15-H33
1.0000
1.098
P58-O61
1.505(5)
1.489
C16-C17
1.5100(5)
1.531
P58-O62
1.578(6)
1.693
C16-H34
0.9995
1.096
H59-H64
1.005
0.991
C16-H35
0.9997
1.100
O62-H63
1.005
0.971
RMSD
0.065 Å
Bond angles (°)
C2-N1-C13
121.5(1)
129.536
C16-C17- H36
108.84
112.687
C2-N1-H48
119.3
119.047
C16-C17-H37
108.88
111.062
C13-N1-H48
119.25
111.387
N18-C17- H36
108.88
106.346
N1-C2-C3
126.8(2)
123.005
N18-C17-H37
108.83
104.285
N1-C2-C11
115.6(2)
120.246
H36-C17-H37
109.53
107.543
C3-C2-C11
117.6(2)
116.748
C17-N18-C19
105.42(4)
110.081
C2-C3-C4
119.5(2)
120.770
C17-N18-C21
117.16(4)
115.141
C2-C3-H23
120.3
120.656
C17-N18-H50
106.64
106.062
C4-C3-H23
120.2
118.558
C19-N18-C21
113.63(4)
113.264
C3-C4-N5
122.7(2)
121.996
C19-N18-H50
106.67
105.629
C3-C4-H24
118.7
122.029
C21-N18-H50
106.69
105.850
N5-C4-H24
118.6
115.974
N18-C19-C20
111.90(3)
111.886
C4- N5-C6
119.1(2)
121.776
N18-C19-H38
108.87
106.612
C4- N5-H49
120.4
119.692
N18-C19-H39
108.91
107.399
C6-N5-H49
120.5
118.523
C20-C19-H38
108.84
113.272
N5-C6-C7
119.7(2)
119.461
C20-C19-H39
108.80
111.196
N5-C6-C11
119.9(2)
119.233
H38-C19-H39
109.49
106.091
C7-C6-C11
120.3(2)
121.305
C19-C20-H40
109.48
107.585
C6-C7-C8
118.6(2)
118.870
C19-C20-H41
109.46
114.072
C6-C7-H25
120.7
120.497
C19-C20-H42
109.45
111.744
C8-C7-H25
120.7
120.633
H40-C20-H41
109.46
107.490
C7-C8-C9
122.7(2)
121.420
H40-C20-H42
109.46
106.889
C7-C8-Cl
120.4(2)
118.847
H41-C20-H42
109.52
108.727
C9-C8-Cl
117.0(2)
119.733
N18-C21-C22
115.92(3)
114.633
C8-C9-C10
117.8(2)
119.188
N18-C21-H43
107.84
105.833
C8-C9-H26
121.1
122.471
N18-C21-H44
107.83
106.449
C10-C9-H26
121.1
118.339
C22-C21-H43
107.80
110.976
C9-C10-C11
122.5(2)
121.716
C22-C21-H44
107.84
111.021
C9-C10-H27
118.8
116.140
H43-C21-H44
109.51
107.523
C11-C10-H27
118.7
122.143
C21-C22-H45
109.47
107.878
C2-C11-C6
121.3(2)
119.448
C21-C22-H46
109.43
113.139
C2-C11-C10
120.7(2)
123.065
C21-C22-H47
109.47
111.979
C6-C11-C10
118.1(2)
117.483
H45-C22-H47
109.52
108.829
N1-C13-C14
112.18(5)
113.090
H45-C22-H47
109.47
107.896
N1-C13-C15
114.14(5)
114.908
H46-C22-H47
109.47
106.971
N1-C13-H23
105.70
102.804
N1-H48-O53
109.7(4)
160.205
C14-C13-C15
112.50(4)
112.432
O52-P51-O53
109.6(4)
118.326
C14-C13-H23
105.71
105.282
O52-P51-O54
110.7(4)
112.552
C15-C13-H23
105.77
107.166
O52-P51-O55
107.7(3)
108.699
C13-C14-H30
109.45
108.617
O53-P51-O54
108.0(3)
109.174
C13-C14-H30
109.49
114.029
O53-P51-O55
111.0(3)
102.543
C13-C14-H31
109.53
110.151
O54-P51-O55
109.4
104.137
H29-C14-H30
109.45
107.772
H48-O53-P51
109.5
141.744
H29-C14-H31
109.46
107.456
H48-O53-H64
109.5(3)
96.870
H30-C14-H31
109.44
108.592
P51-O53-H64
118.544
113.169
C13-C15-C16
116.02(4)
116.850
P51-O54-H57
109.434
112.759
C13-C15-H32
107.78
105.350
P51-O55-H56
109.45
106.393
C13-C15-H33
107.78
111.163
O54-H57-O60
152.62
172.312
C16-C15-H32
107.81
108.709
H59-P58-O60
109.47
106.240
C16-C15-H33
107.77
108.080
H59-P58- O61
106.8(4)
111.947
H32-C15-H33
109.57
106.135
H59-P58-O62
108.7(4)
100.693
C15-C16-C17
110.09(3)
112.212
O60- P58-O61
111.1(4)
124.278
C15-C16-H34
109.28
109.383
O60- P58-O62
108.7(3)
104.611
C15-C16-H35
109.31
106.991
O61-P58-O62
112.02
106.329
C17-C16-H34
109.35
110.830
P58- H59-H64
109.5
109.330
C17-C16-H35
109.31
110.727
H57-O60-P58
112.0(4)
119.982
H34-C16-H35
109.49
106.464
P58-O60-H63
109.5
104.281
C16-C17-N18
111.86(4)
114.339
O53-H64-H59
161.56
162.347
RMSD
3.382°
3.2 Frontier orbitals and quantum chemical calculations
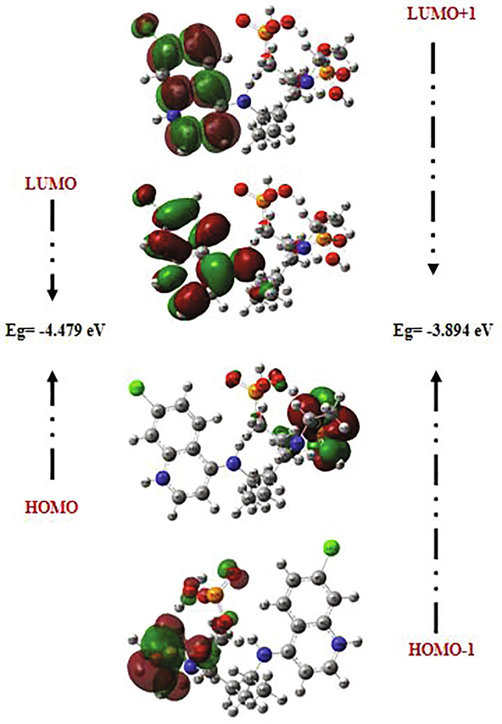
Frontier molecular orbitals (FMOs) often play dominant roles in molecular systems. The fundamental idea of this theory can be abridged in the form of a simple rule telling the condition for a simple course of the reaction by the requirement of the maximal positive overlap between LUMO (empty state) and HOMO (filled state) orbitals. LUMO (lowest unoccupied molecular orbital) is directly related to electron affinity, while HOMO (highest occupied molecular orbital) is related to ionization potential (Xavier and Periandy, 2015; Abraham et al., 2017). These orbitals help to understand the chemical stability and the reactivity of the molecule (Asiri et al., 2011; Kosar, 2011). In order to predict the energetic behaviors and the reactivity of the chloroquine and the chloroquine phosphate against COVID-19 virus, the FMOs in the electronic transitions and their energies difference Eg are determined. A detailed analysis of the HOMOs and LUMOs orbitals is listed in Table 4, where orbital energies, energy band gap and reactivity descriptors (like electron affinity, chemical softness, ionization potential, chemical softness….) are reported. The gap between two energetic states describes the molecular chemical reactivity. The energies of the four important FMOs (HOMO, HOMO − 1, LUMO and LUMO + 1) were calculated via the TD-DFT approach with B3LYP/6-31G* level. Their 3D plots are illustrated in Figs. 3 and 4. It is clear from the figure of the chloroquine molecule that the HOMO and LUMO orbitals are localized essentially on the benzene and pyridine rings. The green color represents the negative phase; on the other hand the red color corresponds to the positive phase which is well clarified in the density of states (DOS) spectrum (Fig. 5). DOS spectrums characterize the energy levels per unit energy increment and its composing in energy. The displaying study per orbital shows that the green and the red lines in these curves correspond to the HOMO and LUMO energy levels, respectively. As a result, the energy level of the HOMO orbital is about −5.594 eV and the energy level of the LUMO orbital is about −1.115 eV. The HOMO-LUMO gap energy (Eg) of the chloroquine is equal to −4.479 eV. This low energy value promotes the transfer of electrons in the chloroquine molecule. These values are compatible with those obtained by the DOS spectrum. The state HOMO-1 form another set of degenerate orbital −5.747 eV lower in energy than the HOMO set. As shown for the chloroquine phosphate, LUMO orbital lying at −2.59 eV, located on all the atoms of the benzene and pyridine rings. The HOMO orbital is lying at −5.228 eV. Consequently, Eg is closed to −2.629 eV. The change observed here in the gap value from −4.479 eV to −2.629 eV in solution involves an expected high reactivity for the chloroquine phosphate. This decrease in gap energy makes the flow of electrons easier, so the molecule becomes soft and more reactive. We can also note that the chloroquine molecule is harder before adding the phosphate groups, given the energy value of gap. This result is in agreement with the strong increase in the dipole moment value of 6.05 Debye (of chloroquine) to 24.49 Debye (of chloroquine phosphate). I = –EHOMO, A = –ELUMO, η = (I–A)/2, ζ = 1/2η, χ = (I + A)/2, μ = –(I + A)/2, ω = μ2/2η and ΔNmax. = –μ/η.
Parameters
Chloroquine
Chloroquine phosphate
ELUMO
−1.115
−2.599
EHOMO
−5.594
−5.228
EHOMO-ELUMO
−4.479
−2.629
ELUMO+1
−0.375
−1.579
EHOMO-1
−5.747
−5.473
EHOMO-1- ELUMO+1
−5.372
−3.894
Reactivity descriptors
Ionization potential (I)
5.594
5.228
Electron affinity (A)
1.115
2.599
Chemical hardness (η)
2.239
2.629
Chemical softness (ζ)
1.1195
1.3145
Electronegativity (χ)
3.3545
3.9135
Chemical potential
−3.3545
−3.9135
Electrophilicity index (ω)
2.512
2.912
Maximum charge transfer index
1.498
1.488
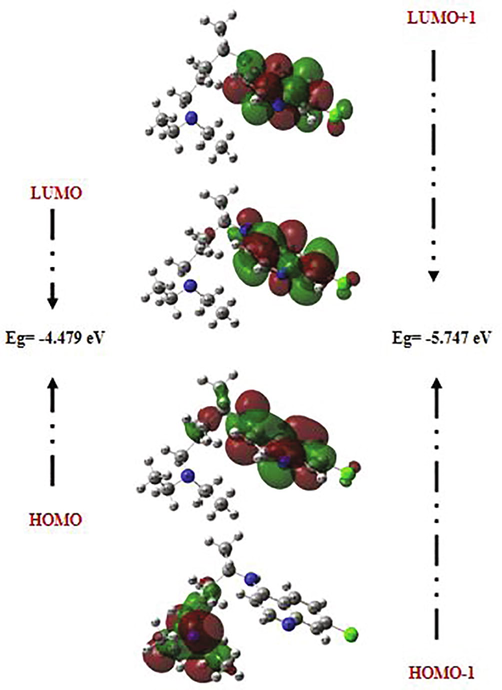
The atomic orbital compositions of the HOMO, HOMO-1, LUMO and LUMO + 1 frontier molecular orbitals for chloroquine molecule.
The atomic orbital compositions of the HOMO, HOMO-1, LUMO and LUMO + 1 frontier molecular orbitals for chloroquine phosphate.
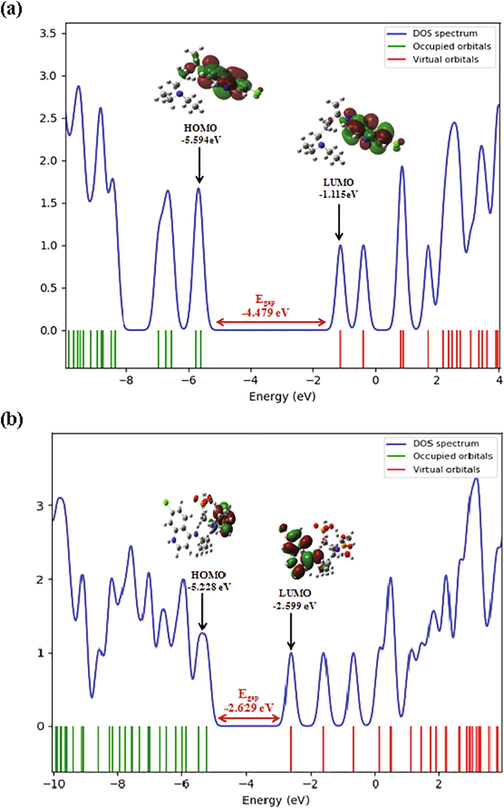
DOS spectrum of chloroquine (a) and chloroquine phosphate (b) molecules.
Using the energies of FMOs, we calculated the reactivity descriptors of chloroquine and chloroquine phosphate molecules. A = − ELUMO: represent the electron affinity; I = − EHOMO represent the ionization potential and μ = 1/2(I + A) is the electronic chemical potential. The chemical hardness (η) is found to be 2.239 and 2.629 eV for chloroquine and chloroquine phosphate, respectively. The chemical softness (ζ) has been computed and found to be 1.1195 and 1.3145 eV−1. Moreover, the electrophilicity index (ω) is about 2.512 eV for chlroquine and 2.912 eV for chloroquine phosphate. Based on the value found of the electrophilicity index, we can conclude that the chloroquine phosphate is a good electrophile better than chloroquine. Therefore, it is able to accept an electron doublet in order to form bonds with another reagent which is necessarily a nucleophile. Electronegativity is also determined (χ = (I + A)/2) and it is found to be χ chloroquine = 3.3545 eV and χ chloroquine phosphate = 3.9135 eV.
3.3 Molecular electrostatic potential
The molecular electrostatic potential (MEP) is a well-established tool for the study of molecular reactive properties and to describe intermolecular interactions (Reed and Weinhold, 1985). It allows us to search the most reactive nucleophilic and electrophilic sites of a molecule against the reactive biological potentials (Gökce et al., 2013). These sites promote the formation of hydrogen bonds. The electrophilic site indicates a strong attraction, while the nucleophilic site indicates a strong repulsion. The electrostatic potential diagrams of chloroquine and chloroquine phosphate are illustrated in Fig. 6 at B3LYP/6-31G* method. MEP diagram gives negative, positive and neutral electrostatic potential regions in terms of color grading and is an indicator in the research of molecular structure properties. The red color represents the most electronegative electrostatic potential. That is, atoms in this region have a tendency to attract electrons (electrophilic). The blue color indicates the most electropositive potential (strong attraction) and the red color indicates the most electronegative potential (strong repulsion). Regions where the potentials are zero are denoted by green color. As a results, MEP surfaces varies between −5.504 0.10−2 a.u (deepest red) to 5.504 0.10−2 a.u (deepest blue) for chloroquine and between −0.116 a.u to 0.116 a.u for chloroquine phosphate. As can be seen, the MEP map of chloroquine molecule (Fig. 6a), a maximum positive region is localized on the nitrogen N3 and hydrogen H30 atoms indicating a possible site for electrophilic attack. The zero potential sites (green color) are found in the benzene ring. For the chloroquine phosphate (Fig. 6b), the positive potential (blue and light blue) sites are found in the benzene and pyridine rings (electrophilic reactivity). It can be inferred that the oxygen atoms O61 and O62 indicate the neutral potential of the molecule.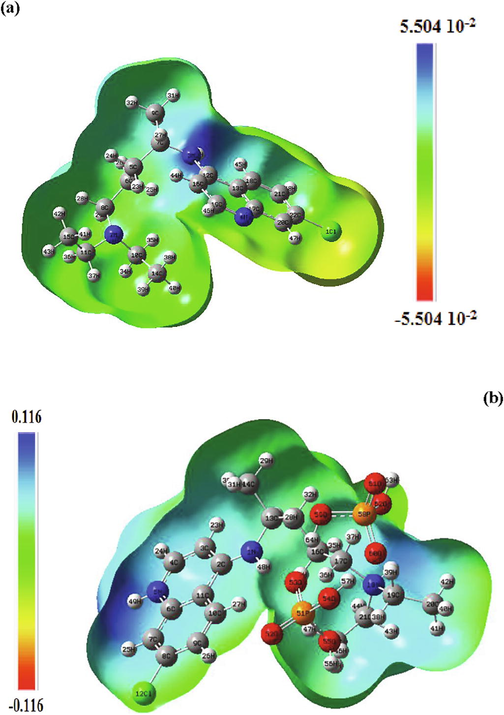
Molecular electrostatic potential (MEP) maps of chloroquine and chloroquine phosphate molecules.
3.4 Molecular docking analysis
Molecular docking studies of chloroquine and chloroquine phosphate ligands were carried out with four structures of COVID-19 protein (PDB ID: 6 M03, 5R7Y, 5R81 and 6LU7). The two ligands were tested for drug-likeliness properties. Calculations were performed using the iGEMDOCK program through the generic evolutionary method (GA) and an empirical scoring function. Both ligands and target proteins structures were adapted with Discover Studio Visualizer software. All crystallographic water molecules were removed.
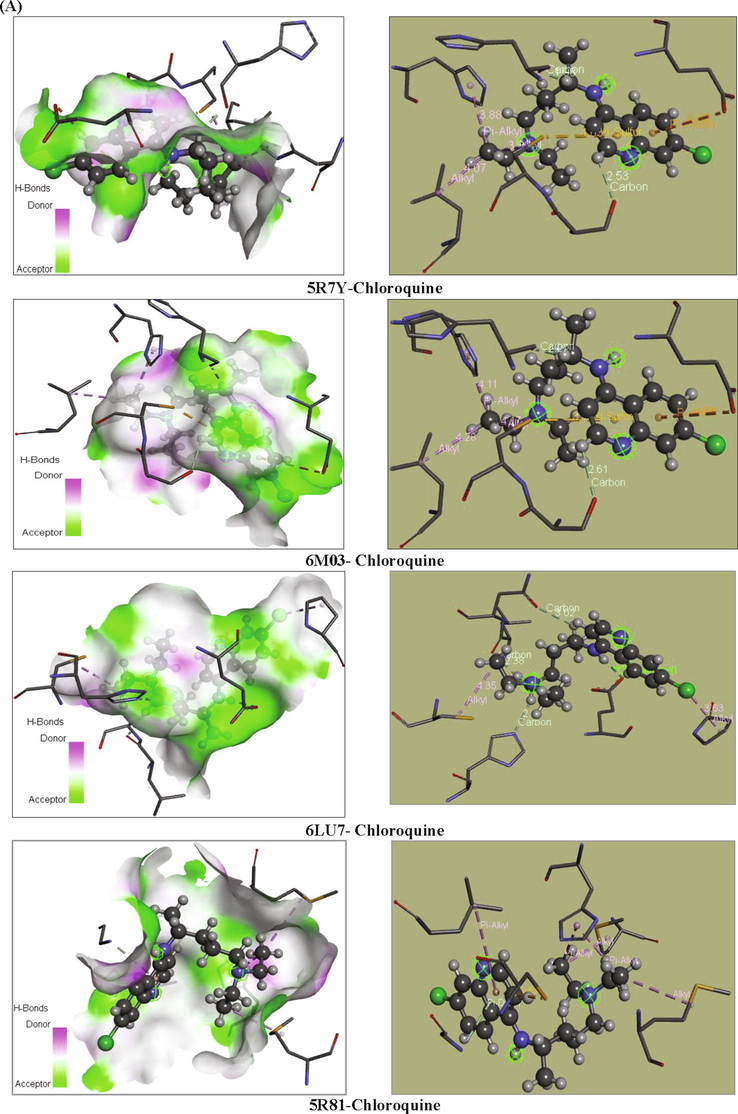
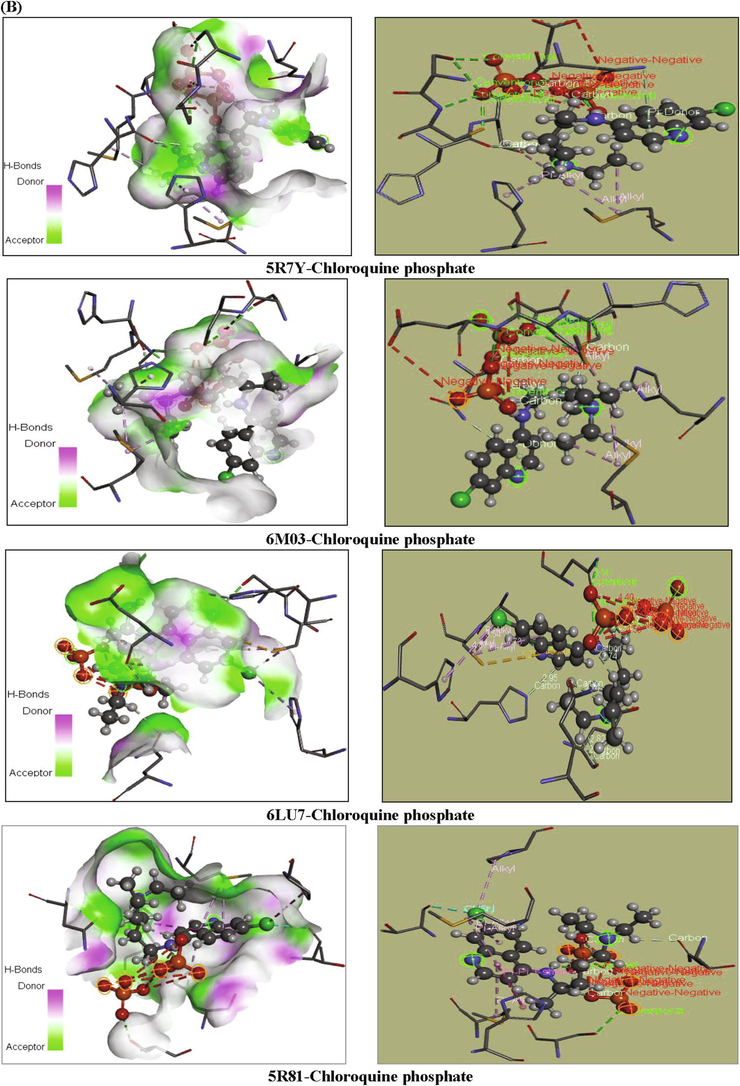
Our goal is to determine the modes of interaction of protein-ligand complexes while looking for favorable orientations for the binding of a ligand to a receptor (Duhovny et al., 2002; Seeliger and de Groot, 2010; Amin et al., 2010; Ahmed et al., 2013; Ghalla et al., 2018). In our case, the receptor represents the COVID-19 protein which has one or more specific active sites, more or less accessible. At each step, the interactions are affected and the best pose of the ligands is determined. 10 poses have been obtained; we have chosen the best pose with the lowest energy. These best poses, as presented in Fig. 7, were selected for investigating the different types of interactions that introduce a biological signal.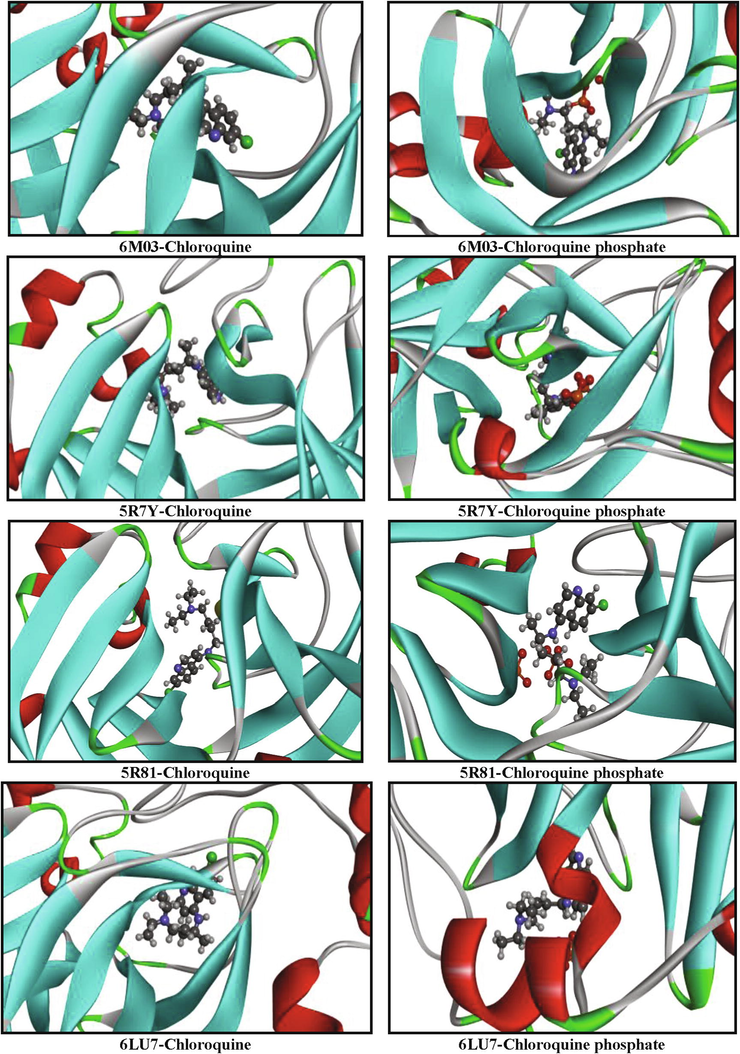
Orientation of chloroquine and chloroquine phosphate in the active sites of COVID-19 proteins.
3.4.1 Chloroquine
The examination of Table 5 revealed that the chloroquine ligand presented the highest total energy score with the target protein 6 M03 which is equal to −81.866 kcal/mol. Note that the total energy is the sum of the three energies interactions: VDW, hydrogen band and electronic. Van der Waals interaction is a potential energy of attraction between two molecules. It represents the sum of the energies of Keesom, London and Debye. The H-bond represents an interaction between two electronegative atoms. Generally, the energy of an H-bond is of the order of a few tens of KJ/Mol. It varies between 1 and 60 KJ/mol for neutral fragments, and sometimes it can reach higher values for some covalent bonds. The last interaction is electronic; they always take very low values compared to the other two interactions.
Chloroquine
Ligands
6 M03
5R7Y
5R81
6LU7
Total energy
−81.866
−77.498
−68.514
−67.136
VDW
−75.581
−70.605
−65.014
−64.988
H-bond
−6.285
−6.893
−3.500
−2.147
Electronic
0
0
0
0
Affinity
−6.7
−6.6
−6.7
−6.1
Chloroquine phosphate
Ligands
5R7Y
6 M03
5R81
6LU7
Total energy
−99.119
−88.686
−84.817
−82.663
VDW
−66.409
−55.450
−79.862
−69.861
H-bond
−29.499
−30.505
−4.9547
−12.802
Electronic
−3.210
−2.731
0
0
Affinity
−4.5
−3.5
−3.5
−3.6
Chloroquine ligand posses the strongest van der Waals interaction EVDW = -75.581 kcal/mol. The docking pose analysis showed that the chloroquine ligand is oriented with the VDW interactions surrounded by the chains of LEU-141, MET-165, PHE140, HIS163, GLN189, MET49, GLY143, THR25 and VAL42 binding residues in the 6 M03 protein. Also, it have the strongest H-bond interaction EH-bond = -6.893 kcal/mol. The greater negative energy score suggests a more favorable binding mode. Table 6 presents the different interactions between the chloroquine ligand and proteins via the binding residues along with their bond length. Results obtained for protein targets show that the chloroquine ligand has bonded effectively with 6 M03 target sites with two remarkable carbon-hydrogen bond interactions. The mentioned compound is immensely bonded with active residues SER144 (Serine) and HIS164 (Histidine) by carbon-hydrogen bond interactions conduct to more antiviral activity. The first C—H bond interaction was identified between H46 atom and SER144 binding residues and the distance was found to be 2.61 Å. The second C—H bond interaction was identified between H27 and HIS164 with distance 2.27 Å. The hydrogen atom H30 linked to HIS41 amino residues via an alkyl interaction with bond length equal to 4.11 Å. Also, Pi-Sulfur, Pi-Alkyl and Pi-Anion interactions were observed surrounded by the amino acids CYS145, LEU27 and GLU166, having distances 3.99, 4.28 and 4.55 Å, respectively. These results have been well described in Figs. 8 and 9. Furthermore, chloroquine molecule showed total energy score of −77.498 kcal/mol against 5R7Y protein with VDW interaction (−70.605 kcal/mol) and hydrogen bond energy (−6.893 kcal/mol). Regarding the two other proteins (5R81 and 6LU7), the interaction energies are slightly weaker in comparison with the other ligands but as even remain important. The docking calculations led to the following results: the total energies scores are equal to −68.514 kcal/mol and −67.136 kcal/mol for 5R81 and 6LU7, respectively. The van der Waals interactions were found to be EVDW (for 5R81) = −65.014 kcal/mol and EVDW (for 6LU7) = −64.988 kcal/mol. Additionally, the hydrogen bond interactions exhibiting values of −3.500 and −2.147 kcal/mol for 5R81 and 6LU7 receptors. In the chloroquine-5R7Y complex, a Pi-Anion and Pi-Sulfur interactions wrapped by the amino acids GLU166 and CYS145 were formed with bond lengths 4.42 and 4.03 Å. C15 atom made two Alkyl interactions with A:CYS145 and A:LEU27 residues and having distances 3.99 and 4.07 Å. Also, C15 interact with A:HIS41 via a Pi-Alkyl interaction (bond length = 3.88 Å). A:SER144 and A:HIS164 amino residues form two carbon-hydrogen bond interactions with H46 and H27 atoms. Their bonding distances are found to be 2.53 Å and 1.98 Å, respectively. In 5R81virus, A:MET165 and A:MET49 amino residues are involved in the alkyl interaction with C10 and C15 atoms having bond length 4.43 and 3.96 Å. Pyridine group formed Pi-Alkyl, Pi-Sulfur and Pi-Donor hydrogen bond interactions with A:LEU27 (5.13 Å), A:CYS145 (4.08 Å) and A:CYS143 (3.80 Å) residues, respectively. Another Pi-Alkyl interaction is also seen which contributed by A:HIS41 with C15 atom, indicating distance 4.25 Å. For the last ligand 6LU7, the LEU141 (2.38 Å), the ASN142 (3.02 Å) and the HIS163 (2.47 Å) amino acids formed a C—H bond interactions with H29, H27 and H28 atoms of chloroquine. In addition to these weak interactions there are two alkyl interactions; one between PRO168 residues and the Cl atom and the second one is in between CYS145 and the N2 atom, indicating bond distance 3.63 and 4.35 Å, respectively. Subsequently, the H30 atom exhibit a conventional-H bond interaction with GLU166 residues and bonding distance is 2.22 Å.
Ligand
Target protein
Binding residue
Type
Atoms
Bond length (Å)
Interactions
Chloroquine
5R7Y
A:GLU166
A:CYS145
A:CYS145
A:LEU27
A:HIS41
A:SER144
A:HIS164GlutamicAcid
Cysteine
Cysteine
Leucine
Histidine
Serine
HistidineBenzene
Pyridine
C15
C15
C15
H46
H27
4.42
4.03
3.99
4.07
3.88
2.53
1.98Pi-Anion
Pi-Sulfur
Alkyl
Alkyl
Pi-Alkyl
Carbon-H bond
Carbon-H bond
6 M03
A:CYS145
A:GLU166
A:HIS41
A:LEU27
A:SER144
A:HIS164Cysteine
GlutamicAcid
Histidine
Histidine
Serine
HistidinePyridine
Pyridine
H30
H30
H46
H27
3.99
4.55
4.11
4.28
2.61
2.27Pi-Sulfur
Pi-Anion
Alkyl
Pi-Alkyl
Carbon-hydrogen bond
Carbon-hydrogen bond
6LU7
A:LEU141
A:ASN142
A:HIS163
A:PRO168
A:CYS145
A:GLU166Leucine
Asparagine
Histidine
Proline
Cysteine
GlutamicAcidH29
H27
H28
Cl
N2
H30
2.38
3.02
2.47
3.63
4.35
2.22C—H bond
C—H bond
C—H bond
Alkyl
Alkyl
Conventional H-bond
5R81
A:MET165
A:MET49
A:HIS41
A:LEU27
A:CYS145
A:CYS143Methionine
Methionine
Histidine
Histidine
Cysteine
GlutamicAcidC10
C15
C15
Pyridine
Pyridine
Pyridine4.43
3.96
4.25
5.13
4.08
3.80Alkyl
Alkyl
Pi-Alkyl
Pi-Alkyl
Pi-Sulfur
Pi-Donor H-bond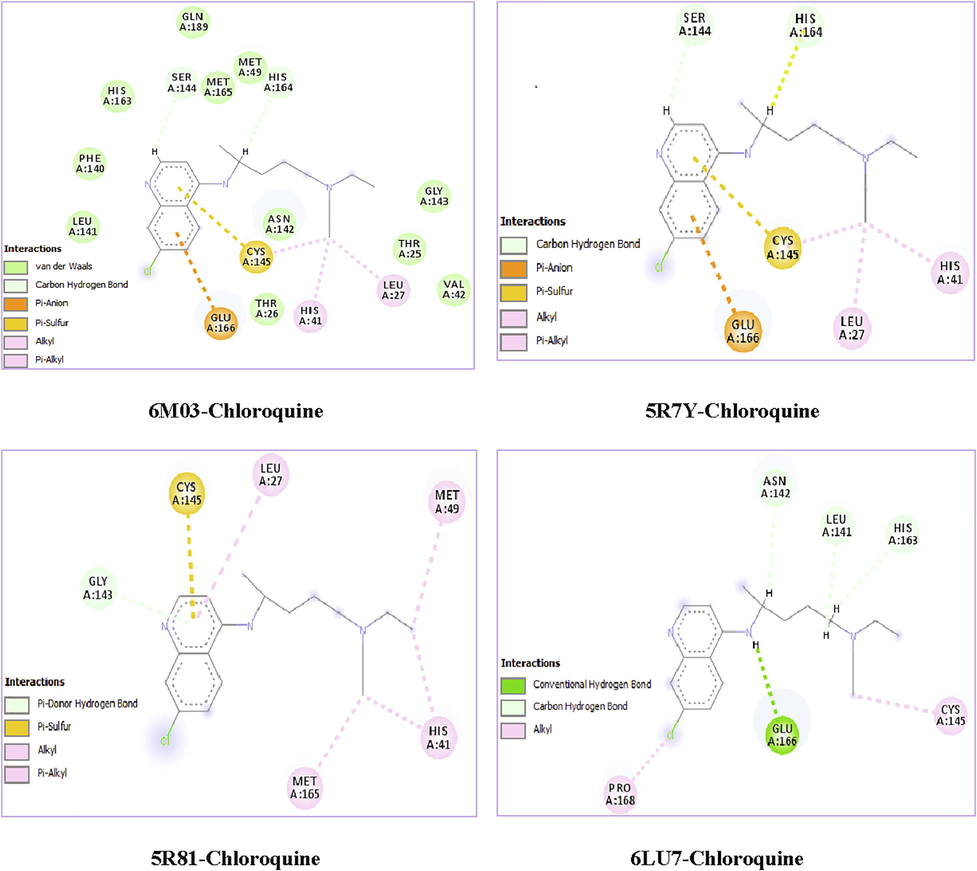
2D visual representations of chloroquine ligand-COVID-19 proteins.
Different interactions between ligand and their receptor.
Different interactions between ligand and their receptor.
In order to upgrade the recognition of the interactions existing between receptor and ligand, the affinities of these complexes were calculated by using AutoDockTools (ADT) (Morris et al., 2008). These affinities describe the strength of a non-covalent interaction between the ligand and its target which binding to a site on its surface. It is premised on the numeral and the nature of the physicochemical interactions. As illustrated in Table 5; the affinities values (in ultimate value) of chloroquine are found to be in the order of 6.7 > 6.6 > 6.1 kcal/mol for (6 M03 and 5R81), 5R7Y and 6LU7, respectively.
3.4.2 Chloroquine phosphate
According to the energetic related results of the docking calculations and the corresponding docking positions, the chloroquine phosphate has better binding interaction with 5R7Y protein (as seen in Table 5 and Fig. 7). This protein strongly interacts with the mentioned ligand, resulting in high inhibition potency. It presented the highest total energy value of −99.119 kcal/mol with a −66.409 kcal/mol van der Waals interaction, also along with important hydrogen and electronic energies equal to −29.499 and −3.210 kcal/mol, respectively. Thereafter, we show that the binding affinities of chloroquine phosphate-6 M03 complex exhibit total energy score equal to −88.686 kcal/mol with EVDW = −55.450 kcal/mol, EH-bond = −30.505 kcal/mol and E electronic = −2.731 kcal/mol. The total energies scores of 5R81 and 6LU7 proteins are found to be −84.817 and −82.663 kcal/mol, respectively. As clearly seen, docking calculations led to the following results: the H-bond interaction equal to −4.954 and −12.802 kcal/mol and their VDW interaction were −79.862 and −69.861 kcal/mol, respectively. For PDB ID: 5R7Y, as shown in Table 7, the amino acid A:MET49 and A:MET165 residues were involved in alkyl interaction with C15 atom with 4.52 and 4.39 Å bond length, respectively. Likewise, C15 atom was linked to A:HIS41 (4.40 Å) throughout pi-alkyl interaction. Moreover, oxygen atom O55 showed a conventional hydrogen bond with amino acid A:GLU166 having distance 2.65 Å. The pyridine group present a Pi-Donor H-bond with A:ASN142, indicating 4.19 Å bond length. For the second 6 M03-chloroquine phosphate complex, A:MET49 interacted with C22 and C20 atoms via alkyl interaction with 3.17 and 4.05 Å bond length. A pi-alkyl interaction was also being formed between A:HIS41 residues and C20 (3.58 Å). In addition, H63 atom (2.45 Å) involve in carbon H-bond with A:HIS164 amino acid. The pyridine ring exhibited pi-donor H-bond interaction with A:ASN142 having 3.79 Å distance. Then, O54 atom has a conventional H-bond interaction with A:GLU166 residues with distance value 3.27 Å. Amino acids A:HIS41 and A:HIS145 forms Pi-Alkyl interactions with Cl atom (4.87 Å) and benzene ring (4.73 Å) for PDB ID: 6LU7. As well, the Cl atom interacts with A:HIS145 via an Alkyl interaction with 3.54 Å distance. The H63 and H24 atoms have a carbon H-bond interactions with A:GLN189 and A:HIS163 residues with distances values 2.74 Å and 2.95 Å, respectively. Finally, the other amino acids A:ASN142 and A:SER144 forms two conventional H-bond interactions with H48 (2.78 Å) and N5 (2.93 Å) atoms. For the last 5R81-chloroquine phosphate complex, an Alkyl interaction was observed between A:PRO168 amino acid residues and Cl atom having 5.02 Å bond length. In addition, two Pi-Alkyl interactions are performed between A:MET165 and A:MET49 residues and pyridine ring. Their bond lengths are equal to 4.40 and 4.67 Å, respectively. A:HIS41, A:THR190 and A:HIS41 amino acid residues interacted with C15, Cl and pyridine ring via Pi-Sigma, halogen and Pi-Pi T shaped interactions, showed distances ranging from 3.04 to 5.01 Å. Chloroquine phosphate present weaker affinities −4.5 kcal.mol−1 (for 5R7Y), −3.6 kcal.mol−1 (6LU7), −3.5 kcal.mol−1 (5R81), −3.5 kcal.mol−1 (6 M03).
-
The results obtained show that the chloroquine penetrates well into the active areas of the protein. Therefore, it can be considered to be a potent inhibitor against COVID-19 diseases. But the chloroquine phosphate molecule showed a better activity rather than chloroquine since it interacts stronger with the receptor. This can be justified by the effect of the addition of the phosphate groups.
| Ligand | Target protein | Binding residue | Type | Atoms | Bond length (Å) | Interactions |
|---|---|---|---|---|---|---|
| Chloroquine phosphate | 5R7Y | A:MET49 A:MET165 A:HIS41 A:GLU166 A:ASN142 |
Methionine Methionine Histidine GlutamicAcid Asparagine |
C15 C15 C15 O55 Pyridine |
4.52 4.39 4.40 2.65 4.19 |
Alkyl Alkyl Pi-Alkyl Conventional H-bond Pi-Donor H-bond |
| 6 M03 | A:MET49 A:MET49 A:HIS41 A:HIS164 A:ASN142 A:GLU166 |
Methionine Methionine Histidine Histidine Asparagine GlutamicAcid |
C22 C20 C20 H63 Pyridine O54 |
3.17 4.05 3.58 2.45 3.79 3.27 |
Alkyl Alkyl Pi-Alkyl Carbon H-bond Pi-Donor H-bond Conventional H-bond |
|
| 6LU7 | A:HIS41 A:HIS145 A:HIS145 A:GLN189 A:HIS163 A:ASN142 A:SER144 |
Histidine Histidine Histidine Glutamine Histidine Asparagine Serine |
Cl Benzene Cl H63 H24 H48 N5 |
4.87 4.73 3.54 2.74 2.95 2.78 2.93 |
Pi-Alkyl Pi-Alkyl Alkyl Carbon-hydrogen bond Carbon-hydrogen bond Conventional H-bond Conventional H-bond |
|
| 5R81 | A:PRO168 A:MET165 A:MET49 A:HIS41 A:THR190 A:HIS41 |
Proline Methionine Methionine Histidine Threonine Histidine |
Cl Pyridine Pyridine C15 Cl Pyridine |
5.02 4.40 4.67 3.81 3.04 5.01 |
Alkyl Pi-Alkyl Pi-Alkyl Pi-Sigma Halogen Pi-Pi T shaped |
3.5 Hybridization effect
Of course, each compound has its own characteristics that distinguish it from the rest. The chloroquine phosphate is initially made up of chloroquine. Evidently, the adding of other atoms in the geometry of the chloroquine has an influence on their stability. The chloroquine compound becomes more stable when adding the phosphate groups since the global minimum energy decreases. Moreover, the smallest dipole moment was obtained for the chloroquine whereas the highest one was obtained for the chloroquine phosphate. This increase shows that the chloroquine is harder before adding the phosphate groups and also it promotes the formation of hydrogen bonds. We also find that by adding phosphate group the gap energy decreases, which involves a high reactivity for the chloroquine phosphate. This decrease in gap energy makes the flow of electrons easier, so the molecule becomes soft and more reactive.
4 Conclusion
Given their high efficiency in the treatment against COVID-19 pandemic, chloroquine derivatives have been studied combining DFT method and molecular docking calculations. The optimized molecular structures of chloroquine and chloroquine phosphate have been carried out using DFT/B3LYP/6-31G* method and their geometrical parameters were also determined. The comparison of the observed and calculated results showed a good agreement. Molecular properties such as frontiers orbitals, gap energies and reactivity descriptors have also been discussed. Results reveal that the addition of the sulfate group resulted in a decrease in the gap energy, which involves an expected high reactivity for the chloroquine phosphate. This decrease in gap energy makes the flow of electrons easier, so the molecule becomes soft and more reactive. The density of states (DOS) was determined and it allowed bettering describing the border orbitals. Thereafter, the calculated MEP maps show the positive potential sites are favorable for nucleophilic attack, whereas the negative potential sites are favorable for the electrophilic attack. Docking results were discussed based on the different interactions between the ligands and proteins. The chloroquine derivatives are found to be a good inhibitor of COVID-19 virus and can, therefore, be effective in controlling this disease. We found that chloroquine phosphate was considered to be the best inhibitor of coronavirus pandemic.
Funding
Researchers supporting project number (RSP-2020/61), King Saud University, Riyadh, Saudi Arabia.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Quantum mechanical, spectroscopic and docking studies of 2-Amino-3-bromo-5-nitropyridine by Density Functional Method. Spectrochim. Acta Part A. 2017;181:153-163.
- [Google Scholar]
- Receptor-and ligand-based study of fullerene analogues: comprehensive computational approach including quantum-chemical, QSAR and molecular docking simulations. Org. Biomol. Chem.. 2013;11:5798-5808.
- [Google Scholar]
- A practical approach to the management of cancer patients during the novel coronavirus disease, (COVID-19) pandemic: an international collaborative group. Oncologist. 2019;25(2020):936.
- [Google Scholar]
- A solid-state dehydration process associated with a significant change in the topology of dihydrogen phosphate chains, established from powder X-ray diffraction. Cryst Growth Des.. 2008;8:3641-3645.
- [CrossRef] [Google Scholar]
- Synthesis, biological evaluation and molecular docking of novel series of spiro [(2H, 3H) quinazoline-2, 1′-cyclohexan]-4 (1H)-one derivatives as anti-inflammatory and analgesic agents. Eur. J. Med. Chem.. 2010;45:2117-2131.
- [Google Scholar]
- Spectrochim. Acta A. 2011;82:444-455.
- Becke’s three parameter hybrid method using the LYP correlation functional. J. Chem. Phys.. 1993;98:5648-5652.
- [Google Scholar]
- J. Chem. Phys.. 1993;98:5648-5652.
- India’s indigenous idea of herd immunity: the solution for COVID-19. Tradit. Med. Res.. 2020;5:182-187.
- [Google Scholar]
- Structure cristallines et moléculaires de trois formes polymorphes de l'oestrone. Acta Crystallogr. B Struct. Cryst. Cryst. Chem.. 1973;29:298-313.
- [CrossRef] [Google Scholar]
- Chloroquine and hydroxychloroquine as available weapons to fight COVID-19. Int. J. Antimicrob. Agents. 2020;105932
- [Google Scholar]
- Severe outcomes among patients with coronavirus disease (COVID-19)—United States. MMWR Morb. Mortal Wkly.. 2019;69(2020):343-346.
- [Google Scholar]
- Epidemiological characteristics of 2143 pediatric patients with 2019 coronavirus disease in China. Pediatrics 2020
- [Google Scholar]
- D. Duhovny, R. Nussinov, H.J. Wolfson, Efficient unbound docking of rigid molecules, (2002).
- Gaussian 09, Revision C.01, Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.;Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H.P.; Izmaylov, A.F.; Bloino, J.; Zheng, G.; Sonnenberg, J.L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J.A., Jr.; Peralta, J.E.; Ogliaro, F.; Bearpark, M.; Heyd, J.J.; Brothers, E.; Kudin, K.N.; Staroverov, V.N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J.C.; Iyengar, S.S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, N.J.; Klene, M.; Knox, J.E.; Cross, J.B.; Bakken, V.;Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R.L.; Morokuma, K.; Zakrzewski, V.G.; Voth, G.A. Salvador, P.; Dannenberg, J.J.; Dapprich, S.; Daniels, A.D.; Farkas, Ö.; Foresman, J.B.; Ortiz, J.V. Cioslowski, J.; Fox, D.J. Gaussian, Inc., Wallingford CT (2009).
- GaussView, Guassian, Inc. (Carnergie Office Parck-Building6 Pittsburgh PA 151064 USA), Copyright © 2000-2003 Semichem. Inc.
- Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents. 2020;105949
- [Google Scholar]
- Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents. 2020;105949
- [Google Scholar]
- Intermolecular interactions and molecular docking investigations on 4-methoxybenzaldehyde. Comput. Mater. Sci. 2018;149:291-300.
- [Google Scholar]
- Lett. Org. Chem.. 2013;10:395-441.
- Combined experimental and theoretical studies on the molecular structures, spectroscopy, and inhibitor activity of 3-(2-thienyl) acrylic acid through AIM, NBO, FT-IR, FT-Raman, UV and HOMO-LUMO analyses, and molecular docking. J. Mol. Struct.. 2017;1130:659-668.
- [Google Scholar]
- Experimental, computational, and in silico analysis of (C8H14N2) 2 [CdCl6] compound. J. Mol. Struct.. 2020;128186
- [Google Scholar]
- Spectrochim. Acta A. 2011;78:160-167.
- Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785-789.
- [Google Scholar]
- Chem. Res. Chin. Univ.. 2015;31:597-602.
- G. M. Morris, R. Huey, J. O. Arthur, Using autodock for ligand‐receptor dockingCurr Protoc Bioinformatics. 24 (2008) 8-14.
- Structural, docking and spectroscopic studies of a new piperazine derivative, 1-phenylpiperazine-1,4-diium-bis (hydrogen sulfate) J. Mol. Struct.. 2020;1202:127351
- [Google Scholar]
- Experimental and DFT studies on the molecular structure, spectroscopic properties, and molecular docking of 4-phenylpiperazine-1-ium dihydrogen phosphate. J. Mol. Struct.. 2020;1207:127762
- [Google Scholar]
- A library for package independent computational chemistry algorithms. J. Comput. Chem.. 2008;29:839-845.
- [Google Scholar]
- Absolute hardness: companion parameter to absolute electronegativity. J. Am. Chem. Soc.. 1983;105:7512-7516.
- [Google Scholar]
- Properties and reactivities of niclosamide in different media, a potential antiviral to treatment of COVID-19 by using DFT calculations and molecular docking. Biointerface Res. Appl. Chem.. 2020;10:7295-7328.
- [Google Scholar]
- E. Romano, N. Issaoui, M. E. Manzur, S. A. Brandán, Properties and molecular docking of antiviral to COVID-19 chloroquine combining DFT calculations with SQMFF approach, Inter. J. of Current Adv. Research, Volume 9, Issue 08(A), (2020) 22862-22876.
- Searching potential antiviral candidates for the treatment of the 2019 novel coronavirus based on DFT calculations and molecular docking. Heliyon. 2020;6(8):e04640
- [Google Scholar]
- Molecular docking studies, structural and spectroscopic properties of monomeric and dimeric species of benzofuran-carboxylic acids derivatives: DFT calculations and biological activities. Computat. Biol. Chem.. 2020;107311
- [Google Scholar]
- Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des.. 2010;24:417-422.
- [Google Scholar]
- D.S. Visualizer, Accelrys software inc. Discovery Studio Visualizer. 2 (2005).
- Spectroscopic (FT-IR, FT-Raman, UV and NMR) investigation on 1-phenyl-2-nitropropene by quantum computational calculations. Spectrochim. Acta Part A. 2015;149:216-230.
- [Google Scholar]
- GEMDOCK: a generic evolutionary method for molecular docking Proteins. Struct. Funct. Bioinform.. 2004;55:288-304.
- [Google Scholar]




