

Translate this page into:
Deciphering the interaction of plumbagin with human serum albumin: A combined biophysical and molecular docking study
⁎Corresponding authors. fhusain@ksu.edu.sa (Fohad Mabood Husain), krais@ksu.edu.sa (Rais Ahmad Khan)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
It is important to understand the nature and mechanism of interaction of drugs with serum albumins as such interactions govern their pharmacokinetics and pharmacodynamics. Plumbagin is a natural phytocompound, mainly present in the bark of Plumbaginaceae family plants and it has numerous therapeutic potentials. In this study, the interaction of plumbagin with human serum albumin (HSA) was deciphered using an array of biophysical and computational tools. The UV–vis spectroscopy established the formation of plumbagin-HSA complex with binding constant as 2.32 × 104 M−1. Fluorescence spectroscopy revealed that there was static mode of quenching of HSA’s fluorescence by plumbagin. The binding constant obtained by fluorescence spectroscopy was of the similar order as in case of UV–vis spectroscopy. The negative value of ΔH° (−3.67) indicated an endothermic nature of the reaction and negative value of ΔG° (−6.40 to −6.58) confirmed that complexation of plumbagin to HSA was spontaneous. There was nearly one binding site in HSA for plumbagin. Titration of plumbagin to HSA in presence of site-specific markers showed that plumbagin interacted at sub-domain IIA of HSA, commonly known as Sudlow’s site I. Moreover, the binding resulted in changes in microenvironment of tryptophan while of tyrosine residues remained approximately unchanged. This interaction also resulted in decrease of α-helical content of the studied serum protein. FRET calculations revealed that distance between plumbagin and HSA’s fluorophore (Trp214) was 2.27 nm. Finally, molecular docking studies substantiated the in vitro finding. Plumbagin formed hydrogen bond with Lys199 and Arg257 of HSA. Additionally, Arg222, His242, and Ala261 interacted via van der Waals forces and Leu238, Leu260, Ile264, Ile290, and Ala291 of HSA interacted with plumbagin through hydrophobic forces.
Keywords
Plumbagin
Human serum albumin
Interaction
Molecular docking

- CD
Circular dichroism
- HSA
Human serum albumin
- FRET
Förster resonance energy transfer
Abbreviations
1 Introduction
Human serum albumin (HSA) is a model protein to study the interactions of biological macromolecules with drugs and it does not exhibit toxicity and immunogenicity (Shahabadi and Maghsudi, 2009). HSA performs an array of functions including distribution, deposition of endogenous and exogenous molecules; maintenance of blood’s osmotic pressure; and transportation of drugs to the molecular target (Yasmeen et al., 2017). There are three homologous domains (I, II and III) of HSA which is further subdivided two sub-domains, i.e. A and B. The most commonly characterized sites of HSA known for ligand binding are located in sub-domain IIA and IIIA also referred as Sudlow’s site I and Sudlow’s site II, respectively (Sudlow et al., 1975). Most of the drugs usually interact with HSA via non-covalent interactions. These drugs interact reversibly with the hydroxyl, carboxyl or different binding groups located on the amino acids residues of HSA via hydrogen bond, van der Waal’s forces, hydrophobic interactions, and ionic interactions (Zia et al., 2018). Drugs showing weak binding affinity to the carrier protein have short life-span in serum and their distribution is poor; while drugs exhibiting greater binding affinity are relatively more efficacious (Hall et al., 2013). Hence, the nature and strength of interaction of drugs with serum albumins is a vital parameter to be considered to study as it determines its pharmacodynamics and pharmacokinetics (Sułkowska, 2002). The information regarding the binding strength, number of binding sites, structural alterations induced in protein due to interaction yield useful details about the clearence and pharmacological distribution of the drugs.
Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone), belonging to naphthoquinone family of compounds, is an important member of quinone family which is found in leaf, stem bark, and root of many plant families such as Dioncophyllaceae, Plumbaginaceae, Ebenaceae, and Ancistrocladaceae. The phytocompound is reported to exhibit anticancer, antioxidant, anti-inflammatory, antibacterial, hypolipidemic, antifungal, and neuroprotective activities (Liu et al., 2017). The therapeutic application of plumbagin is limited mainly due to the low solubility in aqueous solutions. The nanoformulation based on drug carrier systems may be a novel approach to enhance the bioavailability and thereby increasing the clinical application (Bothiraja et al., 2013). Researchers have developed many drug carrier systems for the effective delivery of plumbagin for improved anticancer efficacy (Duraipandy et al., 2014).
The present study focuses on the elucidation of binding mode of plumbagin with HSA that was achieved using numerous biophysical and computational tools. The UV–vis spectroscopy confirmed the plumbagin-HSA complex formation. Fluorescence quenching study was performed to obtain the quenching mode and values of various thermodynamic parameters. Site-specific marker experiment was done to get the exact binding site of plumbagin in HSA. Circular dichroism analysis was carried out to study the alterations in the secondary structural content of the studied serum protein. Finally, molecular docking studies substantiated the in vitro finding and provided detailed information regarding the interacting amino acids.
2 Materials and methods
2.1 Materials
Human serum albumin (HSA) free from fatty acids (A3782), warfarin (258016), ibuprofen (I110), and plumbagin (P7262) were procured from Sigma-Aldrich, USA. All other reagents and chemicals used in this present study were checked for analytical grade before using.
2.2 Preparation of stock solutions
The solution of HSA (300 μM) was prepared in phosphate buffer (10 mM, pH 7.4). The stock solution (1 mM) of plumbagin was prepared in DMSO. The solution of warfarin and ibuprofen (6 mM each) was made in HPLC grade ethanol. The dilution of the stock solutions was made in phosphate buffer otherwise stated.
2.3 UV–visible spectroscopy
The UV–vis spectroscopy was carried out using UV-2600 spectrophotometer, Shimadzu, Japan. Briefly, an increasing concentration of plumbagin (3–24 μM) was consecutively added to a fixed concentration (7.5 µM) of the human serum albumin (HSA). All spectral recordings were taken at 298 K and dilutions were made in phosphate buffer. The baseline correction was performed using phosphate buffer. All the absorption spectra were recorded in 200–400 nm range.
2.4 Steady state fluorescence
The fluorescence emission spectra of the samples were recorded using Shimadzu 5301-PC spectrofluorometer, Japan, in quartz cuvette of 1.0 cm path length. The excitation of HSA and plumbagin-HSA complex was done at 280 nm and emission spectra were obtained in 290–600 nm. HSA (3 µM) concentration was fixed and a varying concentration (0.5–4.0 μM) of plumbagin was titrated. The experiment was performed at three different temperatures to quantify the values of various thermodynamic parameters.
2.5 Competitive displacement assay
To obtain the precise site of binding of a molecule in a protein, certain markers of known site-specific binding are used. In competitive displacement assay, 6 µM cite marker was added to 3 µM HSA to saturate all the binding sites present in HSA. The site marker-HSA mixture was excited at 280 nm and emission spectra were recorded in the absence and presence of plumbagin (0.5–4.0 μM) in 290–550 nm range. This experiment was done separately for ibuprofen and warfarin as mentioned previously (Ishtikhar et al., 2014b).
2.6 Assessment of ionic interactions
This assay was performed to elucidate whether the ionic interactions are involved in the formation of HSA-plumbagin complex. Fluorescence emission signal of HSA (3 µM) was recorded in the absence and presence (0.5–4.0 μM) of plumbagin by exciting at 280 nm in phosphate buffer. The emission spectra was recorded form 290 to 550 m. The experiment was repeated at varying concentrations (0, 50, and 100 mM) of NaCl.
2.7 Synchronous fluorescence studies
Synchronous fluorescence assay was performed to find out the alterations in the microenvironment of aromatic amino acids (mainly tryptophan and tyrosine). In this experiment, the difference between emission and excitation wavelengths (Δλ) are kept fixed. A varying concentration of plumbagin (5–40 μM) was added to the fixed amount of HSA (3 µM). The Δλ = 15 nm was set for tyrosine residue in which excitation and emission wavelengths were set from 240 and 255 nm, respectively. Likewise, Δλ = 60 nm was fixed tryptophan residue where the excitation and emission wavelengths were 240 nm and 300 nm.
2.8 Circular dichroism (CD) measurements
The CD spectra of HSA and HSA-plumbagin complex were recorded using J-815 JASCO spectropolarimeter. The spectra of free HSA (7 µM), HSA-plumbagin complex (1:1 and 1:2 molar ratios) were recorded at 25 °C using thermostatically controlled cell holder. The spectra in this study are the average of three scans. One mm quartz cuvette was used in far-UV CD analysis.
2.9 Förster resonance energy transfer (FRET)
For FRET, fluorescence emission signal of HSA (10 µM) and absorption spectrum of plumbagin (10 µM) were recorded. The spectral overlap was calculated using PhotochemCAD™ software. The normalized overlap of absorption and emission spectrum were used to for the calculation of critical distance and energy transfer efficiency (E) as per the Förster’s theory (Forster and Sinanoglu, 1965).
2.10 Three-dimensional fluorescence spectroscopy
All 3D fluorescence spectra were recorded using Shimadzu spectrofluorophotometer RF-6000, Japan. The 3D fluorescence emission signals of HSA (5 μM) and HSA-plumbagin complex (1:2 molar ratio) were recorded at room temperature. The emission and excitation wavelengths were 200–500 nm and 200–400 nm, respectively.
2.11 Molecular docking
The in silico interaction between HSA and plumbagin was performed by using AutoDock Vina (Trott and Olson, 2009). AutoDock-vina performs faster and is also known to do more accurate calculations than AutoDock 4. The structure of HSA was obtained from Protein Data Bank (www.rcsb.org) [PDB ID: 1AO6]. The water molecules in the coordinate file of HSA were deleted using MGL Tools-1.5.4 (Morris et al., 1998). The merging of non-polar hydrogen atoms was performed along with the addition of Kollman charges to the receptor molecule. The grid centre was fixed to x = 22.717, y = 33.725, and z = 38.864 with grid size as 52 × 48 × 48 Å. The receptor molecule was then saved in pdbqt format. The three-dimensional conformer of plumbagin [CID: 10205] was obtained from PubChem website in SDF format and converted to PDB format using Chimera 1.14. The roots of ligand molecule were detected using MGL Tools to make it flexible and the coordinate was then saved in pdbqt format. The other parameters of docking were left as default and post docking assessment was done using Discovery Studio, PyMol 2.2.3, and LigPlot+. The solvent accessible surface areas (ASA) of HSA and plumbagin-HSA complex were obtained from online server (http://cib.cf.ocha.ac.jp/bitool/ASA) available from Center for Informational Biology, Ochanomizu University.
3 Results and discussion
3.1 UV–vis spectroscopy
UV–vis absorption spectroscopy is an important tool to evaluate the changes induced in protein by the interaction of ligand. The absorption spectra of the protein in the absence and presence of plumbagin is shown in Fig. 1A. The characteristic absorption profile of HSA exhibits two absorption maxima, one at 218 nm and other at 280 nm. The absorption maxima near to 220 nm is due to the framework conformation of HSA while the peak near 280 nm is due to the π → π* transition of aromatic amino acids viz. Tyr, Phe, and Trp (Mohammadi et al., 2017). The successive addition of plumbagin resulted in decrease in absorption at 218 nm along with the red shift of approximately 14 nm. This change indicates the formation of complex between HSA and plumbagin that leads to the conformational alterations in the framework of HSA. On contrary, the peak at 280 nm slightly increased indicating a slight change on the conformation of aromatic amino acids due to the interaction of plumbagin (Lou et al., 2017).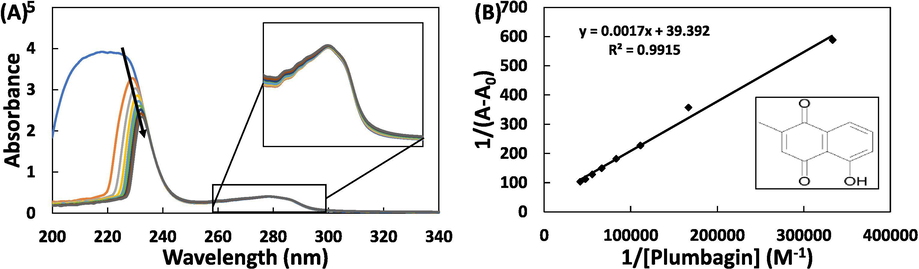
(A) UV–vis absorption spectra of HSA in the absence and presence of different concentrations of plumbagin, enlarged portion of 260–300 nm (inset); (B) double inverse plot, structure of plumbagin (inset). The concentration of HSA and plumbagin was 7.5 µM and 3–24 μM respectively.
The quantitative analysis of UV–vis spectroscopic data was performed using equation-1 (Qais et al., 2016):
3.2 Steady state fluorescence
The fluorescence spectroscopy was deployed to assess the binding of plumbagin with HSA. The fluorescence emission spectra of HSA and HSA-plumbagin complexes is shown in Fig. 2A. It is clear from the data that HSA exhibited emission maximum at 335 nm when excited at 280 nm. The titration of plumbagin to HSA resulted in a progressive fluorescence quenching of the protein with a blue shift to 321 nm. This florescence quenching and blue shift caused by plumbagin indicates the formation of HSA-plumbagin complex. The quantitative analysis of fluorescence emission data was done using Stern–Volmer equation-3 (AlAjmi et al., 2020):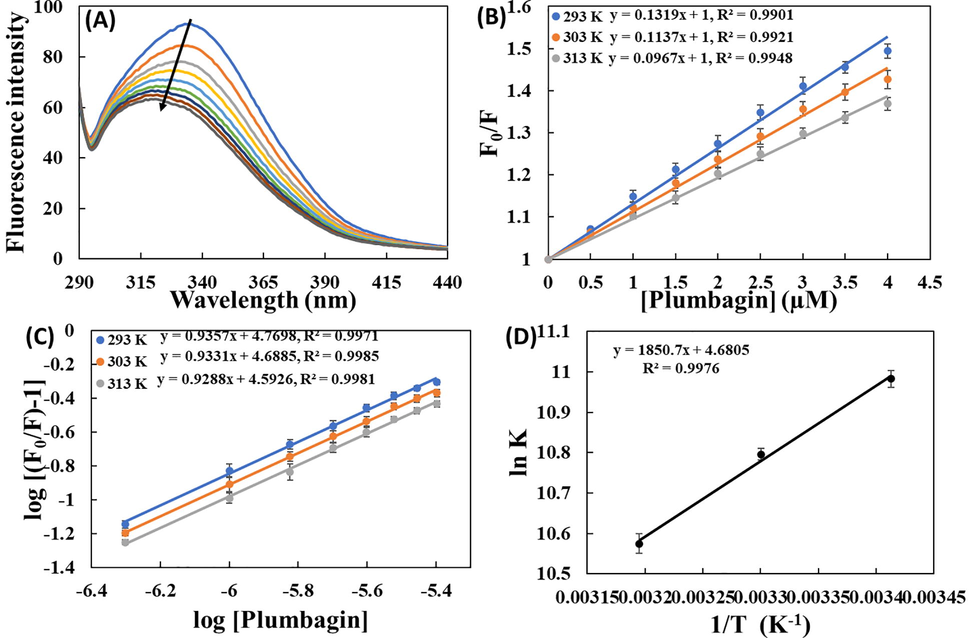
(A) Fluorescence emission signals of HSA and HSA-plumbagin complex at varying concentrations; (B) Stern–Volmer plots for the interaction of plumbagin with HSA at varying temperatures; (C) Double log plot for interaction of plumbagin with HSA; (D) Van’t Hoff plot. The concentration of HSA and plumbagin was 3 µM and 0.5–4.0 μM respectively.
Temp (K)
Ksv (×104 M−1)
Kq (×1013 M−1 s−1)
R2
K (×104 M−1)
n
293
13.12 ± 0.57
2.28 ± 0.09
0.9901
5.93 ± 0.94
0.935 ± 0.016
303
11.51 ± 0.39
1.99 ± 0.06
0.9921
4.90 ± 0.54
0.933 ± 0.009
313
09.67 ± 0.45
1.67 ± 0.07
0.9948
3.96 ± 0.77
0.928 ± 0.014
3.3 Determination of thermodynamic and binding parameters for plumbagin and HSA interaction
The values of the number of binding sites (n) and binding constants (K) were obtained using the equation-5 (Nusrat et al., 2018):
where, K is binding constant; and n is number of binding sites. The slope of double log plot yielded the values of n and y-axis intercept gave the values of binding constants (Fig. 2C). The values of K were found to be 5.93 × 104, 4.90 × 104, and 3.96 × 104 M−1 at 298, 303, and 313 K, respectively. The n remained nearly one indicating that plumbagin preferentially has one binding site in HSA. Binding constant and n obtained at three temperatures is mentioned in Table 1. The data makes it obvious that the binding constant decreased with increasing temperature indicating that HSA-plumbagin complex was relatively more stable at lower temperature compared to physiological temperature.
The values of binding constant at different temperatures were used for the assessment of thermodynamics of the HSA-plumbagin complex formation. In case of drug-protein interactions, non-covalent forces such as van der Waals forces, hydrophobic interactions, hydrogen bonds and electrostatic interactions are predominant (Khan et al., 2008). The values of thermodynamic parameters and their mathematical sign suggest the nature forces responsible for the binding. The values of enthalpy and entropy change were obtained from van’t Hoff’s plot using equation-6:
pH
Temp (K)
ΔG (kcal mol−1)
ΔH (kcal mol−1)
TΔS (kcal mol−1)
R2
7.4
293
−6.40 ± 0.38
2.72 ± 0.13
0.9876
303
−6.49 ± 0.32
−3.67 ± 0.21
2.81 ± 0.14
313
−6.58 ± 0.39
2.91 ± 0.14
The value of Gibb’s free energy change (ΔG°) was obtained using equation-7 (Abdel-Rahman et al., 2019):
The ΔG° was obtained in −6.40 to −6.58 kcal mol−1 range. Its negative value shows that the binding of plumbagin to HSA was spontaneous reaction (Li et al., 2007).
3.4 Evaluation of binding site of plumbagin in HSA
It has been deciphered from HSA’s crystal structure that there are 3 homologous domains named I, II, and III. Each one of these domains is divided into two sub-domains i.e. A and B. To obtain site of binding of drug molecules in HSA, some site-markers with known binding site are used. Crystallographic studies have confirmed that warfarin binds at Sudlow’s site I (sub-domain IIA) and ibuprofen binds at Sudlow’s site II (sub-domain IIIA) of HSA (Sudlow et al., 1975). Stern–Volmer plot was made for comparative assessment of binding of plumbagin in the absence and presence of site markers using equation-3 as shown in Fig. 3A. The KSV values obtained in site-specific marker experiment is enlisted in Table 3. The data showed that KSV value slightly changed in presence of ibuprofen indicating that plumbagin did not compete for Sudlow’s site II and hence have different binding site. On contrary, KSV value remarkably decreased when warfarin was present with HSA. The finding illustrates that plumbagin has competence for binding site as that of warfarin. Moreover, a comparison of the titration results was plotted in terms of relative fluorescence intensity (Fig. 3B). A small change in fluorescence quenching behaviour was observed in presence of ibuprofen. However, there was relatively larger change in fluorescence quenching behaviour of HSA in presence of warfarin suggesting that binding site of plumbagin was same as that of warfarin. Site-marker experiment confirmed that plumbagin interacted at sub-domain IIA of HSA.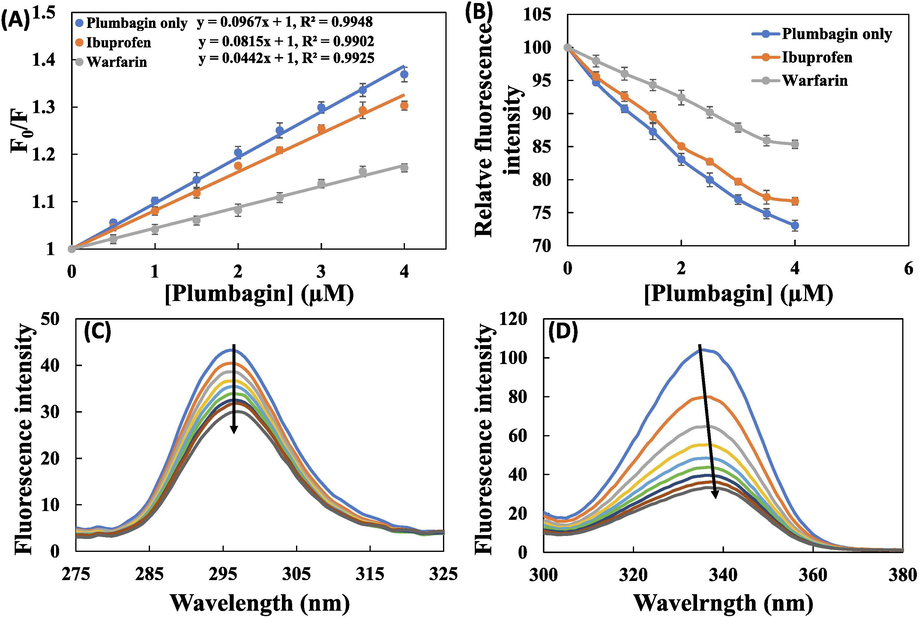
(A) Stern–Volmer plot for plumbagin-HSA interaction in the absence and presence of different site markers; (B) Plots showing plumbagin induced quenching of HSA’s fluorescence in presence of site markers; (C) Synchronous fluorescence spectra of human serum albumin HSA (3 µM) in the absence and presence of plumbagin (5–40 μM) at Δλ = 15 nm; (D) Synchronous fluorescence spectra of human serum albumin HSA (3 µM) in the absence and presence of plumbagin (5–40 μM) at Δλ = 60.
Site Marker
Ksv (×104 M−1)
R2
No site marker
9.67 ± 0.45
0.9948
Ibuprofen
8.14 ± 0.29
0.9902
Warfarin
4.41 ± 0.32
0.9925
3.5 Synchronous fluorescence spectroscopy
The steady state fluorescence data only gives information about the binding constant and various thermodynamic parameters. Synchronous fluorescence analysis data gives information regarding the changes in molecular environment of fluorophores of HSA caused by the binding of drugs (Ibrahim et al., 2010). Any shift (blue shift or red shift) in the fluorescence emission maximum gives valuable information about the alterations of polarity around the fluorophore of HSA (Akram et al., 2020). There is concurrent excitation and emission in synchronous fluorescence, in which their difference (Δλ) is kept constant. Δλ at 60 nm gives information regarding the changes in the microenvironment of tryptophan, whereas Δλ = 15 nm gives information about the microenvironment of tyrosine (Miller, 1979).
The synchronous fluorescence signals of HSA and HSA-plumbagin complex is shown in Fig. 3C and D. There was a negligible shift in the fluorescence emission maxima of HSA at Δλ = 15 (Fig. 3C). This indicates that microenvironment around tyrosine was not changed remarkably. However, at Δλ = 60 nm (Fig. 3D), there was 2 nm red shift showing that polarity in the vicinity of tryptophan residue was increased and thereby decreasing the hydrophobicity around this amino acid (Kragh-Hansen, 1981). The emission at 330 nm indicates that tryptophan residue is present near or in close vicinity of non-polar region of HSA (Miller, 1979). The binding of plumbagin has caused the exposure of tryptophan residue to the surrounding water molecules (Bolattin et al., 2016). The synchronous studies deciphered that microenvironment of tryptophan was changed after interaction of plumbagin while of tyrosine remained approximately same.
3.6 Effect of NaCl
This assay gives information about the involvement of ionic interactions in plumbagin-HSA complex formation. The double log plot for HSA’s fluorescence quenching by plumbagin in the presence and absence of NaCl is presented in Fig. 4A. In absence of NaCl, binding constant was obtained as 3.55 × 104 M−1. In presence of 50 and 100 mM NaCl, the binding constant was obtained as 3.51 and 3.49 × 104 M−1 respectively. A negligible alteration in the value of binding constant signifies that there was insignificant involvement of ionic interactions in the binding of plumbagin with HSA.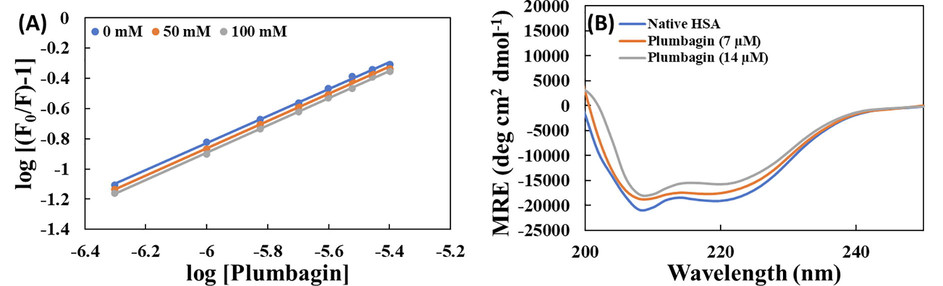
(A) Double plot for the binding of plumbagin with HSA in the absence and presence (50 and 100 mM) of NaCl; (B) Far UV-CD spectra HSA and plumbagin-HSA complex at varying concentrations (1:0, 1:1 and 1:2).
3.7 Circular dichroism spectroscopy
CD is a very sensitive and routinely used tool to study the secondary structure of a protein. The CD spectra of HSA in the absence and presence of plumbagin is shown in Fig. 4B. In a typical CD spectrum of HSA, two negative peaks at 208 and 222 nm are due to the α-helical content of the protein (Zaman et al., 2019; Zhang et al., 2008). CD results are presented in mean residue ellipticity (MRE) which was calculated using equation-8 (Alam et al., 2017):
3.8 3-D fluorescence analysis
Three-dimensional fluorescence spectroscopy provides information about the structural and conformational changes occurred in a protein due to binding of small molecules such as drugs (Siddiqui et al., 2019). The bird's eye view and corresponding contour diagram of HSA in the absence and presence of plumbagin is shown in Supplementary Fig S1. Peak ‘A’ of 3-D fluorescence spectrum of HSA is because of Rayleigh scattering while peak ‘B’ is related to the characteristic of aromatic amino acids (Kabir et al., 2016). The presence of plumbagin decreased the fluorescence intensity of HSA at peak ‘A’ indicating the formation of HSA-plumbagin complex. The fluorescence emission maxima data is presented in Table 4. Moreover, the reduction in fluorescence intensity also depicts that the overall diameter of HSA decreased by the interaction of plumbagin, which in turn caused the reduction of the scattering effect (Siddiqui et al., 2019). The fluorescence signal at peak ‘B’ of the protein also decreased remarkably in presence of plumbagin suggesting the conformational changes around tyrosine and tryptophan residues (Suryawanshi et al., 2016). The results support the findings that the binding of plumbagin with HSA induced changes in the microenvironment of aromatic amino acids and conformational alterations in the protein.
Peak No.
Peak Position λex/λem (nm/nm)
Stokes Shift Δλ (nm)
HSA only
HSA-plumbagin (1:2)
F0
F
F0/F
Peak A
280/330
50
24192.6
21763.0
1.11
Peak B
230/330
100
9493.1
4162.0
2.28
3.9 Förster resonance energy transfer (FRET)
Using FRET, distance between two interacting molecules and energy transfer efficiency could be calculated. For a successful FRET, the fluorescence emission band of donor molecule (HSA) should overlap with the absorption of acceptor (plumbagin). FRET only takes place when; i) the acceptor and donor molecules are in close propinquity, ii) the donor should possess high quantum yield, and iii) there must be proper orientation of transition dipole moment (Xu et al., 2011). If absorption spectrum of plumbagin overlaps substantially with HSA’s emission spectrum, there is higher chances of energy transfer (Forster and Sinanoglu, 1965). The spectral overlap of HSA and plumbagin is presented in Supplementary Fig S2. The degree of energy transfer efficiency is directly correlated the extent of spectral overlap. Energy transfer efficiency was calculated using equation-10 (Il’ichev et al., 2002):
The values of spectral overlap and critical distance was obtained as 2.64 × 10−15 cm3 L mol−1 and 1.96 nm respectively. The distance between Trp214 of HSA and plumbagin was obtained as 2.27 nm with efficiency of energy transfer as 29.04%. The values obtained in FRET using above-mentioned equations are enlisted in Supplementary Table S2. According to Förster’s theory, the maximum R0 value lies between 5 and 10 nm and the maximum r value ranges from 7 to 10 nm (Chen et al., 1990). The obtained values suggested a high probability of energy transfer from HSA to plumbagin. Moreover, these values also indicate that there was static quenching mode of energy transfer that caused an obvious reduction in fluorescence intensity of HSA (Chi et al., 2010).
3.10 Molecular docking
In silico binding analysis tools such as molecular docking and simulation play an important role in the development of new drugs as these tools elucidate the binding site of drugs to biological macromolecules (Rohs, 2005). The advantage of such techniques are that it takes lesser time and provide a thorough information about the interaction of ligand and receptor both in terms of binding energy and affinity compared to other techniques (Elokely and Doerksen, 2013). Most of times, in silico results corroborates with in vitro and in vivo findings. The structure of HSA was kept rigid while plumbagin was made flexible to attain the energetically most stable conformation. AutoDock Vina yielded nine different binding conformations with different binding energies. The docked conformation having lowest binding energy was used for analysis and discussed in this study. The docked conformation with minimum binding energy is presented in Fig. 5. The binding energy of best conformation was obtained as −7.7 kcal mol−1 which corroborates with the spectroscopic results. The binding site of plumbagin in HSA was present in sub-domain IIA of HSA which further corroborates the site-marker studies. Plumbagin formed two hydrogen bonds with Lys199 and Arg257 at distance of 2.50 and 4.40 Å, respectively. Moreover, Arg222, His242, and Ala261 interacted via van der Waals forces. Leu238, Leu260, Ile264, Ile290, and Ala291 of HSA interacted with plumbagin through hydrophobic forces. The details of the interaction are given in Table 5. The in silico results corroborated the experimental results and strengthened our findings.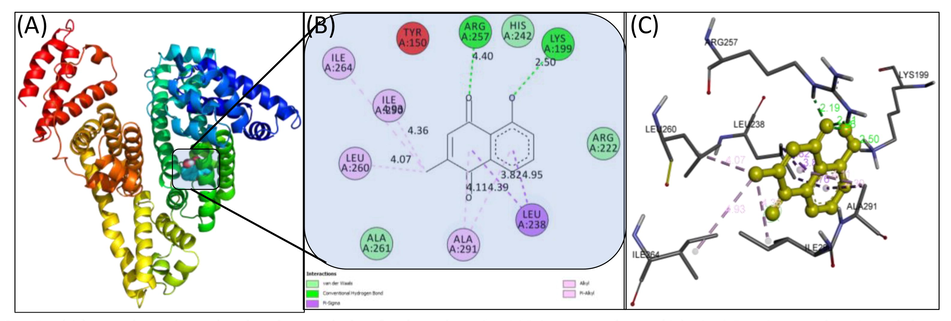
(A) Molecular docked model of plumbagin-HSA complex; (B) A 2D view obtained by Discovery Studio; (C) Plumbagin is shown with interacting amino acids of HSA.
S. No.
Amino acid
Distance (A°)
Nature of interaction
sub-domain
1.
Lys199
2.50
Hydrogen bond
IIA
2.
Arg257
4.40
Hydrogen bond
IIA
3.
Leu238
Hydrophobic interaction
IIA
4.
Leu260
Hydrophobic interaction
IIA
5.
Ile264
Hydrophobic interaction
IIA
6.
Ile290
Hydrophobic interaction
IIA
7.
Ala291
Hydrophobic interaction
IIA
8.
Arg222,
Van der Waal forces
IIA
9.
His242
Van der Waal forces
IIA
10.
Ala261
Van der Waal forces
IIA
The ASA of free HSA was found to be 54957.69 Å2. The binding of plumbagin to HSA reduced the total ASA to 54775.7 Å2 further validating the interaction of plumbagin to HSA. The maximum decrease in ASA of the interacting amino acids were Ala291, Leu238, Lys199, Ile290, Arg222, Leu260, and Arg257 by 33.42, 30.07, 18.30, 12.97, 11.93, 10.89, and 10.13 Å2 respectively as shown in Supplementary Table S3.
4 Conclusion
The findings of this study provide useful information regarding the interaction of plumbagin with HSA. There was progressive quenching of fluorescence of HSA by plumbagin. The binding constant obtained by fluorescence spectroscopy ranged from 3.96 to 5.93 × 104 M−1. The negative value of ΔH° suggested that the binding of plumbagin to HSA was endothermic reaction. Moreover, ΔG° value established that the binding process was spontaneous in nature and thermodynamically favourable. The molecular docking study deciphered that the forces responsible complexation were hydrogen bonds, hydrophobic interactions, and van der Waals forces. There was overall decrease in the solvent accessible surface area of HSA after the binding. These findings are significant to understand the nature interaction of plumbagin with HSA at molecular level and will assist in biomedical and pharmaceutical research.
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at the KSU for funding this work through research group project number RG-1440-059. FAQ is thankful to CSIR, New Delhi, India [File no. 09/112(0626)2k19 EMR] for providing senior research fellowship (SRF).
Conflict of interest
All the authors declare that there is no conflict of interest.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Development, structural investigation, DNA binding, antimicrobial screening and anticancer activities of two novel quari-dentate VO(II) and Mn (II) mononuclear complexes. J. King Saud Univ. - Sci.. 2019;31:52-60.
- [CrossRef] [Google Scholar]
- Binding of cationic Cm-E2O-Cm gemini surfactants with human serum albumin and the role of β-cyclodextrin. J. Mol. Liq.. 2020;312:113365.
- [CrossRef] [Google Scholar]
- Understanding the interaction between α-1-acid glycoprotein (AGP) and potential Cu/Zn metallo-drugs of benzimidazole derived organic motifs: A multi-spectroscopic and molecular docking study. Spectrochim. Acta Part A Mol. Biomol. Spectrosc.. 2020;225:117457.
- [CrossRef] [Google Scholar]
- Multi-spectroscopic and molecular modelling approach to investigate the interaction of riboflavin with human serum albumin. J. Biomol. Struct. Dyn. 2017
- [CrossRef] [Google Scholar]
- Titanium dioxide nanoparticles preferentially bind in subdomains IB, IIA of HSA and minor groove of DNA. J. Biomol. Struct. Dyn.. 2017;36:2530-2542.
- [CrossRef] [Google Scholar]
- Systematic investigation on the interaction of bovine serum albumin with ZnO nanoparticles using fluorescence spectroscopy. Colloids Surf. B Biointerfaces. 2013;102:257-264.
- [CrossRef] [Google Scholar]
- Interaction of hydralazine with human serum albumin and effect of β-cyclodextrin on binding: insights from spectroscopic and molecular docking techniques. Ind. Eng. Chem. Res.. 2016;55:5454-5464.
- [CrossRef] [Google Scholar]
- Improved pharmaceutical properties of surface modified bioactive plumbagin crystals. Int. J. Surf. Sci. Eng.. 2013;7:181.
- [CrossRef] [Google Scholar]
- Chen, G., Huang, X., Xu, J., Wang, Z., Zheng, Z., 1990. No Title, in: Method of Fluorescent Analysis. Science Press, Beijing, pp. 2–122.
- Binding of oxytetracycline to bovine serum albumin: spectroscopic and molecular modeling investigations. J. Agric. Food Chem.. 2010;58:10262-10269.
- [CrossRef] [Google Scholar]
- Cyril, L., Earl, J., Sperry, W., 1961. No Title, in: Biochemists Handbook. E & FN Epon Led. Press, London, p. 83.
- Duraipandy, N., Lakra, R., Kunnavakkam Vinjimur, S., Samanta, D., K, P.S., Kiran, M.S., 2014. Caging of plumbagin on silver nanoparticles imparts selectivity and sensitivity to plumbagin for targeted cancer cell apoptosis. Metallomics 6, 2025–2033. https://doi.org/10.1039/C4MT00165F.
- Docking challenge: protein sampling and molecular docking performance. J. Chem. Inf. Model.. 2013;53:1934-1945.
- [CrossRef] [Google Scholar]
- Forster, T., Sinanoglu, O., 1965. No Title, in: Modern Quantum Chemistry. Academic Press, New York, pp. 93–137.
- The study on the interaction between human serum albumin and a new reagent with antitumour activity by spectrophotometric methods. J. Photochem. Photobiol. A Chem.. 2004;167:213-221.
- [CrossRef] [Google Scholar]
- Automated ligand- and structure-based protocol for in silico prediction of human serum albumin binding. J. Chem. Inf. Model.. 2013;53:907-922.
- [CrossRef] [Google Scholar]
- Interactions between antimalarial indolone-N-oxide derivatives and human serum albumin. Biomacromolecules. 2010;11:3341-3351.
- [CrossRef] [Google Scholar]
- Interaction of ochratoxin A with human serum albumin. preferential binding of the dianion and pH effects. J. Phys. Chem. B. 2002;106:452-459.
- [CrossRef] [Google Scholar]
- Effect of glycation on human serum albumin–zinc interaction: a biophysical study. J. Biol. Inorg. Chem.. 2018;23:447-458.
- [CrossRef] [Google Scholar]
- Interaction of the 5-fluorouracil analog 5-fluoro-2′-deoxyuridine with ‘N’ and ‘B’ isoforms of human serum albumin: a spectroscopic and calorimetric study. Mol. BioSyst.. 2014;10:2954-2964.
- [CrossRef] [Google Scholar]
- Interaction of 5-fluoro-5’-deoxyuridine with human serum albumin under physiological and non-physiological condition: a biophysical investigation. Colloids Surf. B Biointerfaces. 2014;123:469-477.
- [CrossRef] [Google Scholar]
- Interaction of a tyrosine kinase inhibitor, vandetanib with human serum albumin as studied by fluorescence quenching and molecular docking. J. Biomol. Struct. Dyn.. 2016;34:1693-1704.
- [CrossRef] [Google Scholar]
- Mechanistic inhibition of non-enzymatic glycation and aldose reductase activity by naringenin: binding, enzyme kinetics and molecular docking analysis. Int. J. Biol. Macromol.. 2020;159:87-97.
- [CrossRef] [Google Scholar]
- Interaction of mitoxantrone with human serum albumin: spectroscopic and molecular modeling studies. Eur. J. Pharm. Sci.. 2008;35:371-382.
- [CrossRef] [Google Scholar]
- In-vitro binding analysis of bovine serum albumin with sulindac/chlorpromazine: spectroscopic, calorimetric and computational approaches. J. Mol. Liq. 2019:112124.
- [CrossRef] [Google Scholar]
- Molecular aspects of ligand binding to serum albumin. Pharmacol. Rev.. 1981;33:17-53.
- [Google Scholar]
- Interaction of rhein with human serum albumin investigation by optical spectroscopic technique and modeling studies. Biochim. Biophys. Acta - Proteins Proteomics. 2007;1774:51-58.
- [CrossRef] [Google Scholar]
- Anticancer properties and pharmaceutical applications of plumbagin: a review. Am. J. Chin. Med.. 2017;45:423-441.
- [CrossRef] [Google Scholar]
- Characterizing the binding interaction of fungicide boscalid with bovine serum albumin (BSA): a spectroscopic study in combination with molecular docking approach. J. Photochem. Photobiol. B Biol.. 2017;173:589-597.
- [CrossRef] [Google Scholar]
- Recent advances in molecular luminescence analysis. Proc. Anal. Div. Chem. Soc.. 1979;16:203-208.
- [Google Scholar]
- The effect of dichlorvos on the structural alteration of serum albumins: a combined spectroscopic and molecular dynamic simulation approach. Monatshefte für Chemie - Chem. Mon.. 2017;148:1141-1151.
- [CrossRef] [Google Scholar]
- Morris, G.M., Goodsell, D.S., Halliday, R.S., Huey, R., Hart, W.E., Belew, R.K., Olson, A.J., 1998. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 19, 1639–1662. https://doi.org/10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B.
- Interaction of catecholamine precursor l-Dopa with lysozyme: a biophysical insight. Int. J. Biol. Macromol.. 2018;109:1132-1139.
- [CrossRef] [Google Scholar]
- Understanding the mechanism of non-enzymatic glycation inhibition by cinnamic acid: an in vitro interaction and molecular modelling study. RSC Adv.. 2016;6:65322-65337.
- [CrossRef] [Google Scholar]
- Molecular flexibility in ab initio drug docking to DNA: binding-site and binding-mode transitions in all-atom Monte Carlo simulations. Nucleic Acids Res.. 2005;33:7048-7057.
- [CrossRef] [Google Scholar]
- Binding studies of a new copper (II) complex containing mixed aliphatic and aromatic dinitrogen ligands with bovine serum albumin using different instrumental methods. J. Mol. Struct.. 2009;929:193-199.
- [CrossRef] [Google Scholar]
- A comprehensive spectroscopic and computational investigation on the binding of the anti-asthmatic drug triamcinolone with serum albumin. New J. Chem.. 2019;43:4137-4151.
- [CrossRef] [Google Scholar]
- The characterization of two specific drug binding sites on human serum albumin. Mol. Pharmacol.. 1975;11:824-832.
- [Google Scholar]
- Interaction of drugs with bovine and human serum albumin. J. Mol. Struct.. 2002;614:227-232.
- [CrossRef] [Google Scholar]
- Spectroscopic analysis on the binding interaction of biologically active pyrimidine derivative with bovine serum albumin. J. Pharm. Anal.. 2016;6:56-63.
- [CrossRef] [Google Scholar]
- AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem.. 2009;31:455-461.
- [CrossRef] [Google Scholar]
- Interaction of the flavonoid hesperidin with bovine serum albumin: a fluorescence quenching study. J. Lumin.. 2007;126:211-218.
- [CrossRef] [Google Scholar]
- Sensitive and specific DNA detection based on nicking endonuclease-assisted fluorescence resonance energy transfer amplification. J. Phys. Chem. C. 2011;115:16315-16321.
- [CrossRef] [Google Scholar]
- Yasmeen, S., Riyazuddeen, Qais, F.A., 2017. Unraveling the thermodynamics, binding mechanism and conformational changes of HSA with chromolyn sodium: Multispecroscopy, isothermal titration calorimetry and molecular docking studies. Int. J. Biol. Macromol. 105, 92–102. https://doi.org/10.1016/j.ijbiomac.2017.06.122.
- Interaction of anticancer drug pinostrobin with lysozyme: a biophysical and molecular docking approach. J. Biomol. Struct. Dyn.. 2019;37:4338-4344.
- [CrossRef] [Google Scholar]
- Fluorescence study on the interaction of bovine serum albumin with P-aminoazobenzene. J. Fluoresc.. 2008;18:109-118.
- [CrossRef] [Google Scholar]
- Chemotherapeutic drugs and plasma proteins: exploring new dimensions. Curr. Protein Pept. Sci.. 2018;19:937-947.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2020.07.008.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




