

Translate this page into:
Benchmarked molecular docking integrated molecular dynamics stability analysis for prediction of SARS-CoV-2 papain-like protease inhibition by olive secoiridoids
⁎Corresponding author. nchellappan@jazanu.edu.sa (Neelaveni Thangavel)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Objectives
We performed a virtual screening of olive secoiridoids of the OliveNetTM library to predict SARS-CoV-2 PLpro inhibition. Benchmarked molecular docking protocol that evaluated the performance of two docking programs was applied to execute virtual screening. Molecular dynamics stability analysis of the top-ranked olive secoiridoid docked to PLpro was also carried out.
Methods
Benchmarking virtual screening used two freely available docking programs, AutoDock Vina 1.1.2. and AutoDock 4.2.1. for molecular docking of olive secoiridoids to a single PLpro structure. Screening also included benchmark structures of known active and decoy molecules from the DEKOIS 2.0 library. Based on the predicted binding energies, the docking programs ranked the screened molecules. We applied the usual performance evaluation metrices to evaluate the docking programs using the predicted ranks. Molecular dynamics of the top-ranked olive secoiridoid bound to PLpro and computation of MM-GBSA energy using three iterations during the last 50 ps of the analysis of the dynamics in Desmond supported the stability prediction.
Results and discussions
Predictiveness curves suggested that AutoDock Vina has a better predictive ability than AutoDock, although there was a moderate correlation between the active molecules rankings (Kendall's correlation of rank (τ) = 0.581). Interestingly, two same molecules, Demethyloleuropein aglycone, and Oleuroside enriched the top 1 % ranked olive secoiridoids predicted by both programs. Demethyloleuropein aglycone bound to PLpro obtained by docking in AutoDock Vina when analyzed for stability by molecular dynamics simulation for 50 ns displayed an RMSD, RMSF<2 Å, and MM-GBSA energy of −94.54 ± 6.05 kcal/mol indicating good stability. Molecular dynamics also revealed the interactions of Demethyloleuropein aglycone with binding sites 2 and 3 of PLpro, suggesting a potent inhibition. In addition, for 98 % of the simulation time, two phenolic hydroxy groups of Demethyloleuropein aglycone maintained two hydrogen bonds with Asp302 of PLpro, specifying the significance of the groups in receptor binding.
Conclusion
AutoDock Vina retrieved the active molecules accurately and predicted Demethyloleuropein aglycone as the best inhibitor of PLpro. The Arabian diet consisting of olive products rich in secoiridoids benefits from the PLpro inhibition property and reduces the risk of viral infection.
Keywords
Benchmarking docking
Molecular docking
Molecular dynamics
Olive secoiridoids
PLpro
SARS-CoV-2

- AD
-
AutoDock 4.2.1
- ADV
-
AutoDock Vina 1.1.2
- DEKOIS
-
Demanding evaluation kits for objective in-silico screening
- EF
-
Enrichment factor
- g/mol
-
Grams/mole
- kcal/mol
-
Kilocalorie/mole
- MD
-
Molecular dynamics
- MM-GBSA
-
Molecular mechanics generalized Born surface area
- MW
-
Molecular weight
- M
-
Moles
- ns
-
nanoseconds
- OS
-
Olive secoiridoids
- PLpro
-
Papain-like protease
- ps
-
picoseconds
- RMSD
-
Root mean square deviation
- RMSF
-
Root mean square fluctuation
- ROC
-
Receiver operating characteristic curve
- ROC-AUC
-
Area under ROC
- pAUC
-
partial area under ROC
- BEDROC
-
Boltzmann enhanced discrimination of ROC
- RIE
-
Robust initial enhancement
- pTG
-
Partial total gain
- PC
-
Predictiveness curve
- SARS-CoV-2
-
Severe acute respiratory syndrome coronavirus-2
- TG
-
Total gain
Abbreviations
1 Introduction
Virtual screening of a vast number of compounds in freely available databases using molecular docking is one of the frequently applied methods that support the rapid identification of lead compounds. Molecular docking is a computer-aided drug discovery strategy that predicts the conformation of a ligand that exhibits the best binding affinity to a protein. Several docking packages like AutoDock, AutoDock Vina, DOCK, FRED, GemDock, LeDock, rDock, PLANTS, and Smina are freely available (Pagadala et al., 2017). Docking programs vary in their conformation search methods and scoring functions. The docking results on a single target protein may vary with the docking program used. Therefore, it is vital to assess the performance of docking programs on a given target. Performance assessment by benchmarking virtual screening protocols with different docking programs and datasets like DUD-E and DEKOIS 2.0 also resolves artificial enrichment of hits by false positives. These datasets contain validated decoys (inactive molecules); when included in virtual screening, they help recognize an appropriate docking program that can effectively pick up active molecules on top order based on the binding scores placing the decoys on the bottom of the list (Réau et al., 2018). Molecular dynamics (MD) is a computational study that predicts the stability of the docked complexes in a biologically simulated environment and provides insights into the intermolecular interaction mechanisms (Santos et al., 2019).
OliveNetTM is a freely available digital library that provides updated information about the compounds isolated from different parts of the Olea europaea (Oleaceae) tree (Bonvino et al., 2018). Olive phenols are sub-classified into coumarins, flavonoids, glucosides, hydroxybenzoic acids, hydroxycinnamic acids, hydroxy-isochromans, hydroxyphenyl acetic acids, iridoids, lignans, methoxy phenols, phenolic fatty acid esters, secoiridoids, and simple phenols. Olive secoiridoids (OS) form the biggest chemical class of phenolic compounds from different parts of the olive tree. OS are a group of compounds that are esters of phenolic tyrosol/hydroxytyrosol and elenoilc acid or solely derivatives of elenolic acid. Eighty OS has been reported, out of which forty are polyphenolic (hydroxytyrosol derivatives) secoiridoids, and fourteen are phenolic (tyrosol derivative) secoiridoids. The remaining twenty-six secoiridoids are elenolic acid derivatives (Bonvino et al., 2018). OS are the most significant bioactive components of olive and are the main phenolic constituents (70–90 %) of extra virgin olive oil (Lozano-Castellón et al., 2021). Olive oil and olive fruits are inherent in Arabian and Mediterranean diets. The use of olive oil and olive fruits to ameliorate diseases dates back to historical times and is mentioned in the Holy Bible and the Holy Quran. (Ali et al., 2018; Hashmi et al., 2015; Mazzocchi et al., 2019). Recent surveys have corroborated the use of olive oil as adherence to Saudi dietary guidelines and the prevention of cardiovascular diseases and colorectal cancer (Alkhaldy et al., 2019; Azzeh et al., 2017). OS like Ooleuropein, Demethyloleuropein, Oleacein, Oleocanthal, and Ligstroside are abundant in olive oil, olive fruits, and olive leaves (Mazzocchi et al., 2019). OS are associated with the mitigation of oxidative stress, inflammation, diabetes, cancer, coronary heart diseases, hypertension, and the exhibition of immunomodulatory, antimicrobial, and antiviral properties (Castejón et al., 2020; Celano et al., 2019; Emma et al., 2021; Lozano-Castellón et al., 2020; Nediani et al., 2019). Other prominent phenolic compounds in olive leaves, fruits, and oil include p-hydroxybenzoic acid, Gallic acid, Vanillic acid, Apigenin, Rutin, Verbascoside, Protocatechuic acid, Rosmarinic acid, Ferulic acid, Luteolin, Rhamnoliquiritin, and Quercetin that have been already studied by docking and dynamics against several targets of SARS-CoV-2 (Elsbaey et al., 2021; Shawky et al., 2020). Due to the abundance of OS in olives, their pharmacological significance, and the lack of scientific reports on PLpro inhibition properties, they are appropriate for virtual screening against SARS-CoV-2.
Despite several attempts to discover drugs, no study revealed any therapeutic intervention for Coronavirus disease-2019 caused by SARS-CoV-2. Exploring bioactive dietary components for use against SARS-CoV-2 is essential as they present an economically viable and safe therapeutic option. The SARS-CoV-2 genome encodes for a most critical cysteine protease known as PLpro, which plays a vital role in virus replication through cleavage of virus polyproteins Pp1a and Pp1ab. An active triad composed of Cys111, His272, and Asp286 is a characteristic proteolytic site of PLpro (Osipiuk et al., 2021). PLpro also interferes with the host's defense mechanism against virus infection through deubiquitination and deISG15ylation activities (Shin et al., 2020). Hence, PLpro inhibition can block coronavirus replication, and PLpro is a valid target for discovering drugs against SARS-CoV-2.
Here, we describe the virtual screening of the OliveNetTM library by docking secoiridoids to a single SARS-CoV-2 PLpro structure, referred to as PLpro throughout the study. We aimed to benchmark docking using two freely available docking programs, AutoDock Vina 1.1.2 and AutoDock 4.2.1, known actives and decoys from DEKOIS 2.0 database. Besides, MD simulation of the top-ranked secoiriodid docked complex helped predict the stability of the complex. Together, this study aimed to demonstrate a benchmarked molecular docking integrated MD stability analysis for identifying the olive secoiridoid with the potential to inhibit PLpro.
2 Materials and methods
2.1 Virtual screening
2.1.1 PLpro structure preparation
The three-dimensional structure of PLpro (PDB-ID: 6WX4, atomic resolution: 1.66 Å) was downloaded from the Protein Data Bank ( www.rcsbpdb.org) in PDB format. The PDB structure of the protein is a monomer co-crystallized with a non-covalent inhibitor. This structure served as the target for docking and was prepared in the Schrödinger Suite Protein Preparation Wizard before docking (Madhavi Sastry et al., 2013). The inhibitor and water were removed, and the missing residues and side chains were fixed using the Prime module. All hydrogens, including non-polar hydrogens and gasteiger charges, were introduced to the PLpro structure. The partial charges were spread on the deficit atoms of amino acid residues to maintain the uniformity of the system's charge. Finally, all the parameters were saved in PDBQT format suitable for molecular docking.
2.1.2 Benchmarking molecular docking
2.1.2.1 Preparation of OliveNet ligands
The names and 2D structures of 80 OS (Table S1) were obtained from the OliveNet library (https://mccordresearch.com.au). The molecular weights of OS varied between 222.24 and 1077.05 g/mol. The 2D structures of OS were constructed in ChemDraw Ultra 10.0 and loaded in the form of SDF data to the LigPrep tool in the Schrodinger Maestro module for further development (Madhavi Sastry et al., 2013). Epik was chosen, and the area of liquid potential simulations was optimized with an OPLS2005 force field at pH 7.4 for ligand optimization and energy minimization. For docking, a stable conformer with a single protonation state for each ligand was created and saved in PDBQT format. Every ligand was visually inspected for possible high-energy interactions.
2.1.2.2 Preparation of known actives and decoys
The recently included actives and decoys for SARS-CoV-2 PLpro were retrieved from DEKOIS 2.0 library ( http://www.pharmchem.uni-tuebingen.de/dekois/) (Bauer et al., 2013). Twenty-four known actives (Table S2) and 720 decoys (Table S3) are available for benchmarking docking to PLpro (Ibrahim et al., 2020). Known actives serve as a training set and provide a means for assessing the performance of docking programs in the context of their retrieval. Decoys were random and unmatched for the olive secoiridoids (Réau et al., 2018). All known actives and decoys were used without any filter. Actives and decoys structures were downloaded in SDF format from PubChem ( https://pubchem.ncbi.nlm.nih.gov/) and ZINC databases ( https://zinc.docking.org), respectively. A similar ligand preparation to the strategy mentioned earlier under section 2.1.2.1 was applied using the LigPrep tool. One conformation in a single protonated state at pH 7.4 was prepared for each ligand and saved in PDBQT format.
2.1.2.3 Benchmarking molecular docking programs
We used two freely available docking programs, AutoDock Vina 1.1.2 (ADV) (Trott & Olson, 2010) and AutoDock 4.2.1 (AD) (Morris et al., 2009), for benchmarking molecular docking. The binding site to dock ligands to PLpro structure was defined by creating a grid box centered on the catalytic site residue Cys111. The size of the grid box was assigned based on the position of the co-crystallized ligand and to accommodate the large size of OS molecules. A grid box of size 25 × 25 × 25 was selected with grid point spacing 0.375 Å, and dimensions × = 9.2694, y = -20.4856, and z = -37.2115 Å. These dimensions were common for both the docking program runs. The rest of the docking parameters were set at default values in both programs. A total of 821 ligands comprising olive secoiridoids, known actives, and decoys were docked to PLpro in ADV and AD. All rotatable bonds in ligands were allowed to rotate during docking enabling flexible ligand docking. Ten conformations of each compound were analyzed in both programs and conformers with the least binding energy (more negative value) were chosen for performance assessment. The docked complex with the OS predicted as the best inhibitor of PLpro by the best performing program was further analyzed for binding modes in BIOVIA, Dassault Systèmes, Discovery Studio v16, San Diego, 2016.
2.1.2.4 Performance assessment metrics
ADV and AD predicted binding energies of every molecule against PLpro were used to rank them. As known active molecules were involved in screening, it is possible to obtain the OS rank compared to the known molecules. Comparison of performance of different docking programs involves implementing various measures that use these rankings to generate performance scores. Established measures of performance are receiver operating characteristic (ROC) curve, area under ROC (ROC-AUC), partial area under ROC (pAUC), Boltzmann enhanced discrimination of ROC (BEDROC), robust initial enhancement (RIE), enrichment factor (EF), total gain (TG), partial total gain (pTG), and predictiveness curves (PC) (Empereur-Mot et al., 2015). We have calculated all of the known measures, except RIE to quantify and evaluate the performance of the docking programs. Screening Explorer ( http://stats.drugdesign.fr) was used for performing the metrics validation. Screening Explorer is a web-based application for evaluating docking results obtained from different programs (Empereur-Mot et al., 2016). The binding energies for all docked ligands were used as input against an activity tag denoted as 1 for OS, known active molecules, and 0 for decoy molecules. In addition, we have also assessed the ability of the docking programs to retrieve the known actives by using the ranks of known actives against the total number of screened compounds. To evaluate the correlation between the scoring performance of the two docking programs, we applied the statistical measure Kendall's correlation of rank (τ) in Excel.
2.2 Molecular dynamics stability study
The movements of the protein docked to the predicted top inhibitory compound were analyzed and compared with the apo form by MD simulation in the Schrodinger Desmond module (Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2021). The simple point charge model was used, and the force field OPLS2005 was applied. Counter ions were added to neutralize charges (42Na+ and 42Cl- ions), and a 0.15 M NaCl level was maintained to simulate the physiological condition of the human system. At the NPT ensemble temperature of 300 k and 1,01325 bar of pressure strain, the MD simulation was run for 50 ns. The results of MD included RMSD, RMSF, and protein–ligand interactions (Ouassaf et al., 2021). The SigmaPlot (SigmaPlot Version 14.0, Systat Software, San Jose, CA. 2018) program was used for creating comparative graphs.
2.2.1 Binding free energy calculation
Using the Prime-MM-GBSA force field OPLS 2005, the binding free energy measurement for the protein–ligand complex was carried out (Wang et al., 2019). Prime MM-GBSA calculates the binding free energy from contributing energies as follows: ΔGbinding = Gdocked - Gprotein - Gligand.
ΔGbinding = binding free energy of the docked complex; G-docked, G-protein, and G-ligand are the free energies of the docked complex, protein, and ligand, respectively. The results were obtained from 3 iterations during the MD run captured as snapshots from Video S1 and presented as the mean ± standard deviation.
3 Results and discussions
3.1 Virtual screening by benchmarking molecular docking
OS from the OliveNetTM library underwent virtual screening for their potential to inhibit the SARS-CoV-2 PLpro (PDB 6WX4) along with benchmarking known active molecules and decoys from DEKOIS 2.0 database. The ligands were flexible throughout the docking run and moved freely inside the binding sites, and the protein was rigid. Three secoiridoids, Oleuropein pentamer, Oleuropein tetramer, and Oleuropein trimer (MW = 2684.67, 2150.11, and 1613.58 g/mol, respectively), were excluded from the study because of their unfavorable intramolecular interactions during energy minimization. ADV and AD were used for benchmarking the accuracy of prediction, wherein both programs apply empirical free energy scoring. They differ in the search algorithms, where AD uses the Lamarckian genetic algorithm while ADV uses an optimized gradient global and local conformational search (Chang et al., 2010). The binding energies predicted (kcal/mol) by the docking programs were used for active ligand rankings, as shown in Fig. 1.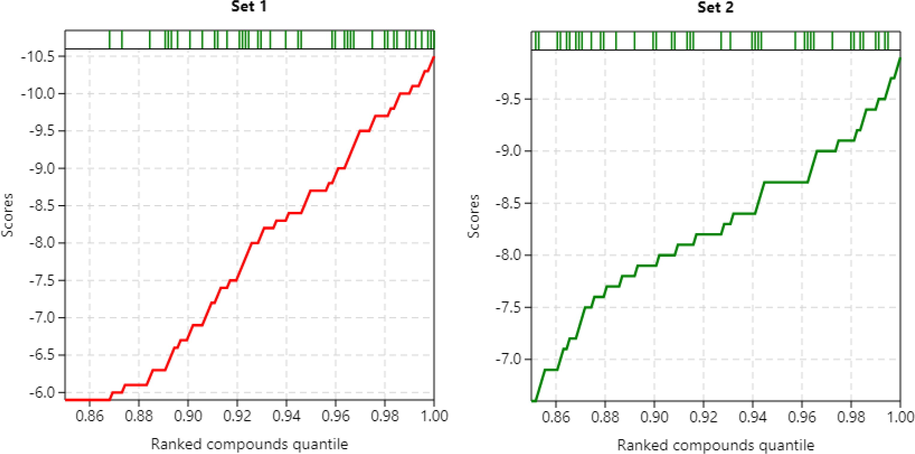
Results of benchmarked docking of olive secoiridoids against SARS-CoV-2 PLpro. Predicted binding energy scores (kcal/mol) plotted against the ranks of the active molecules. (Set 1, 2 = AutoDock Vina, AutoDock). The rug plot above the graph shows the positions of active molecules.
3.2 Performance assessment metrics
The rankings obtained from the docking results were used to construct ROC, EF, and PC curves from which the assessment metrics were calculated. Fig. 2A is the ROC curve at a maximum threshold with ADV ROC-AUC: 0.878; AD ROC-AUC: 0.847 indicating the successful application of scoring functions in differentiating the active molecules from decoys, and both programs have shown relatively equal capacity in rightly ranking the ligands (Empereur-Mot et al., 2015). Fair values of ADV BEDROC: 0.608; AV BEDROC: 0.510 indicate both programs are good in early recognition of active molecules, with ADV showing a marginal improvement in the results (Empereur-Mot et al., 2016). RIE is not reported because BEDROC is the normalization of RIE (Empereur-Mot et al., 2016). TG scores ADV TG: 0.511; AD TG: 0.495, that are above 0.2 suggest a significant relationship between the score variations and active molecules detection. EFmax calculated from ROC of Fig. 2A is 10.35 indicating satisfactory early recognition by both programs. Fig. 2B is the enrichment curve that enables the quantification of early recognition ability by visualization of a true positive fraction over a specific fraction (partial) of the entire ligand set. We analyzed the partial metrics at 1 %: pAUC ADV: 0.588; pAUC AD: 0.505, pTG ADV: 0.684; pTG AV: 0.527, EF1% ADV: 6.47; EF1% AV: 3.88 and partial metrics at 5 % pAUC ADV: 0.558; pAUC AD: 0.499, pTG ADV: 0.545; pTG AV: 0.418, EF5% ADV: 4.66; EF5% AV: 3.62 demonstrating that AutoDock Vina has performed better than AutoDock in predicting the actives. Fig. 2C, PC visually displays the better than random retrieval of actives by both programs, and ADV has a clear edge over AD, with a probability of activity score p(act) at 1 % threshold for ADV: 0.691; p(act) AD: 0545. Fig. 2D displays the ability of the programs to retrieve the known actives: ROC-AUC ADV: 0.905; ROC-AUC AD: 0.895, TG ADV: 0.676; TG AD: 0.655, BEDROC ADV: 0.627; BEDROC AD: 0.610 indicating both programs were equally excellent (Empereur-Mot et al., 2016). The ligand rankings by ADV and AD showed a moderate correlation with Kendall's correlation (τ) = 0.581 (Chang et al., 2010). The ligands placed in the top 1 % rank of the screened molecules were retrieved from Fig. 2B by choosing the set fraction at 0.01 on X-axis. Eight active compounds were retrieved from the 2 docking programs as follows: ADV: OS-3, known actives-2, decoys-3; AD: OS-3, known actives-1, decoys-4. Table 1 shows the enriched top 1 % OS, with two same molecules occupying the list, and Demethyloleuropein aglycone was identified by both software as the best compound with the lowest binding energy (ΔG: ADV = -10.5, AD = -9.9 kcal/mol), hence was chosen for MD analysis. Fig. 3 represents the active conformations of the top 1 % ranked OS bound to PLpro retrieved from AutoDock Vina docking.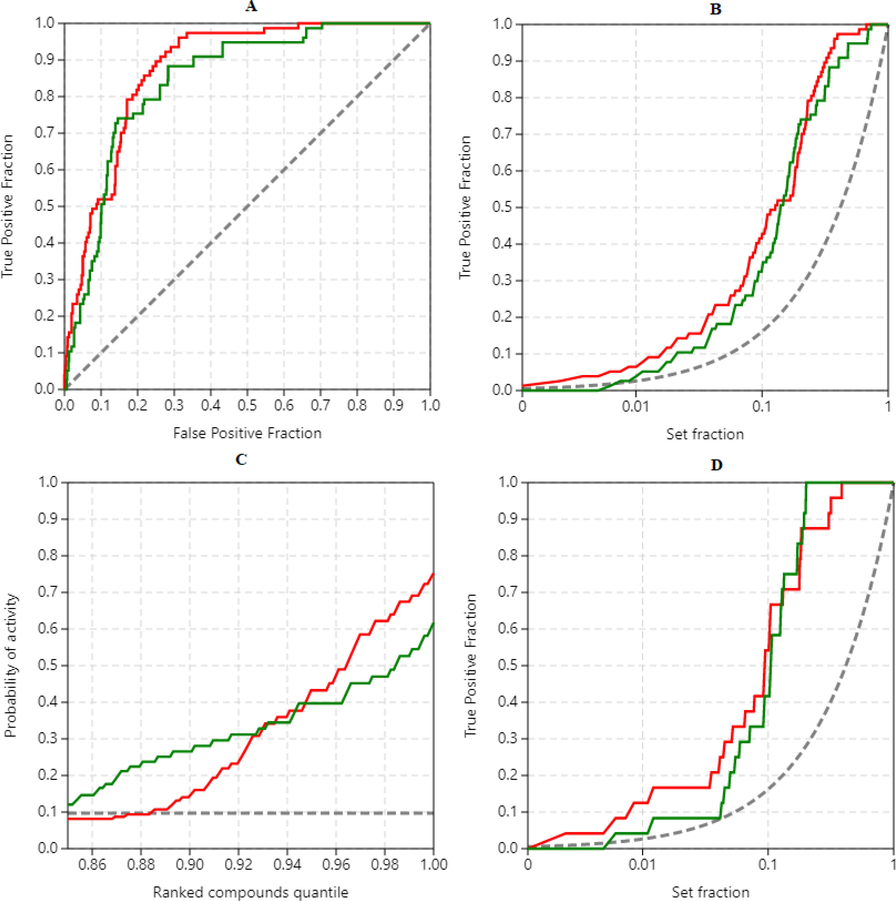
Quantification of docking performance for benchmarking virtual screening of olive secoiridoids as potential SARS-CoV-2 PLpro inhibitors. A) ROC curves B) Enrichment curves for all the screened molecules C) Predictiveness curves D) Enrichment curves for early identification of known active molecules. Red: AutoDock Vina; Green: AutoDock; Dashed lines: Random retrieval.
Olive secoiridoids predicted by AutoDock Vina in the top 1 % rank
Olive secoiridoids predicted by AutoDock in the top 1 % rank
Rank 1
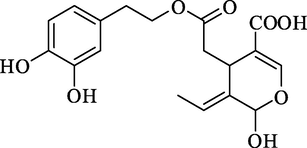
Demethyloleuropein aglyconeRank 4
Demethyloleuropein aglycone
Rank 3
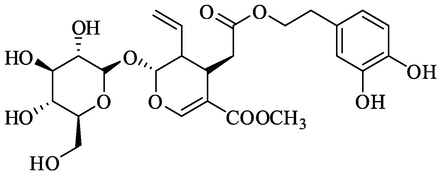
OleurosideRank 5
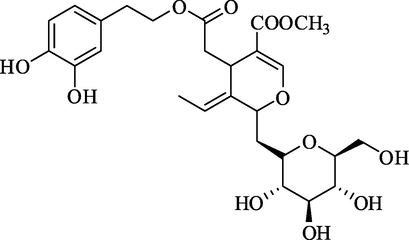
Oleuropein
Rank 6
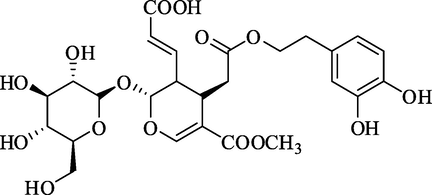
Oleuroside-10-carboxylic acidRank 6
Oleuroside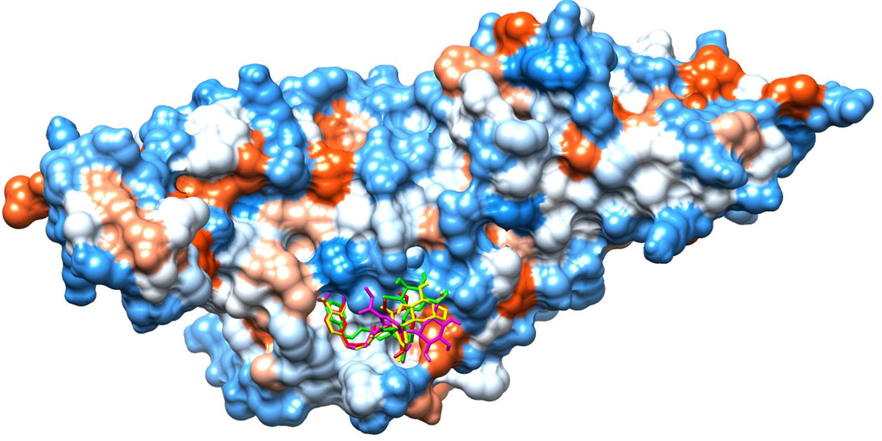
AutoDock Vina retrieved docked poses of the top 1% ranked olive secoiridoids (shown as sticks, red: Demethyloleuropein aglycone, yellow: Oleuroside, magenta: Oleuropein, green: Oleuroside-10-carboxylic acid) inside the binding site of SARS-CoV-2 PLpro (Connolly surface).
3.3 Molecular dynamics stability analysis
Results of MD analysis of Demethyloleuropein aglycone, the top-ranked ligand in complex with PLpro, are shown in Fig. 4, indicating high stability achieved after ligand binding. The RMSD graph (Fig. 4A) exhibits the ligand-bound protein, apo-protein, and ligand conformational changes. The ligand-bound protein reached a stable conformation with a global minimum equal to the apo-protein and maintained stability from 30 ns to 50 ns with RMSD < 2 Å and RMSF < 2.5 Å (Fig. 4B). Low RMSF indicates a less significant fluctuations of the PLpro residues. Fig. 5 is a detailed illustration of the Demethyloleuropein aglycone interactions with PLpro during MD. Arg166 is a positively charged residue located in binding site 2 of PLpro critical for inhibition. Demethyloleuropein aglycone was predicted to form non-covalent electrostatic interactions (67 % of the 50 ns) through the oxygen of phenolic hydroxy moiety, charged interactions through keto oxygen of ester via water bridge (62 %), and Pi-cation interactions (81 %) through the aromatic ring with Arg166, suggesting a potent non-covalent inhibition (Osipiuk et al., 2021). MD results also demonstrated that the ligand occupied site 3 comprising the BL2 loop with Tyr273 and Asp302. Asp302, in a negatively charged state, displayed two strong hydrogen bonds with the H atom of the two phenolic –OH groups for 98 % of the simulation time. Tyr273 exhibited Pi-Pi stacked hydrophobic interactions for 56 % of MD run time. The two phenolic hydroxy groups of Demethyloleuropein aglycone were engaged in hydrogen bonds with Asp302 and Arg166 for a long duration, suggesting that these groups have a significant influence on the nature of binding and stability of interactions with PLpro. Arg166 contributes to PLpro peptidase activity by cleaving the amide linkage between ubiquitin and ISG15 interfering with the human viral defense, thus its interactions with ligand imply its inhibition, delineating the preventative action of OS (Shen et al., 2022). Interestingly the results also demonstrate that the OS occupied the recently identified Pro248-BL2 groove (Shen et al., 2022). Engagement of multiple binding sites leads to cooperative binding resulting in better binding affinity than binding to a single site, justifying potent inhibition (Shen et al., 2022).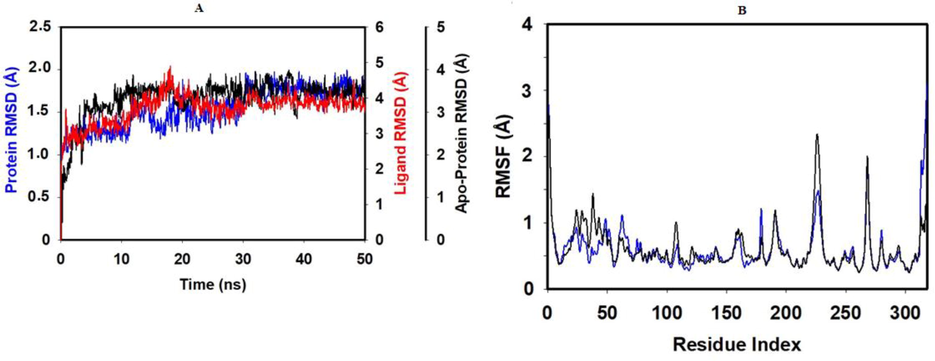
Molecular dynamics stability analysis of Demethyloleuropein aglycone bound SARS-CoV-2 PLpro. A) Comparative RMSD plot B) RMSF of bound PLpro (blue) and apo PLpro (black).
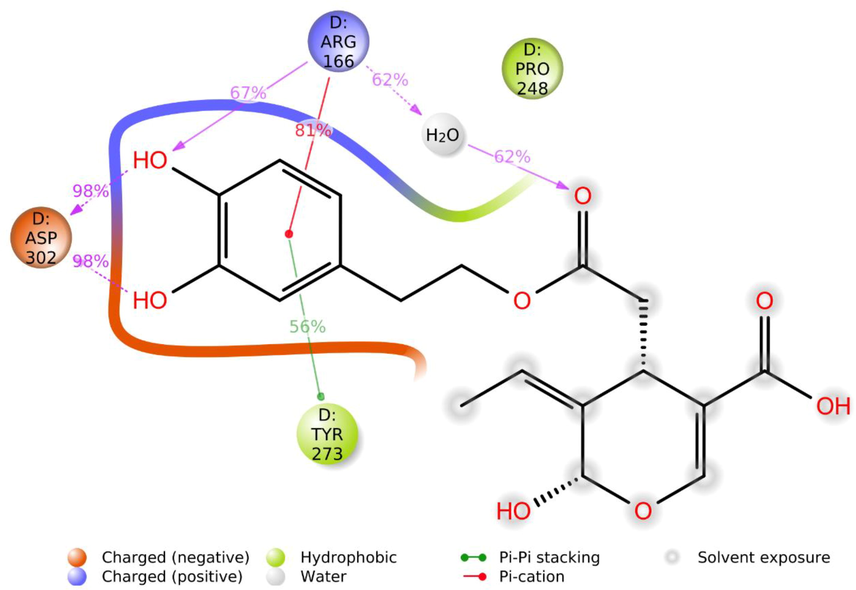
Schematic illustration of Demethyloleuropein aglycone atom interactions with SARS-CoV-2 PLpro residues during molecular dynamics.
3.4 Binding free energy calculation
The binding free energy (MM-GBSA, ΔGbinding, kcal/mol) of Demethyloleuropein aglycone bound PLpro complex was −94.54 ± 6.05 contributed by stabilizing energies: lipophilic energy (ΔGlipo = -16.78 ± 1.23), Van der Waals energy (ΔGvdW = -34.68 ± 2.17), coulomb energy (ΔGcoulomb = –22.56 ± 5.54), and destabilizing energies: generalized Born electrostatic solvation energy (ΔGSolv GB = 22.45 ± 2.78), covalent binding energy (ΔGcovalent = 2.13 ± 1.90). Stabilizing energies contributed more than destabilizing energies suggesting a favorable bound conformation of the protein and ligand (Wang et al., 2019).
4 Conclusion
The benchmarked docking has predicted 4 olive secoiridoids as SARS-CoV-2 PLpro inhibitors at the top 1 % rank out of 821 screened molecules. AutoDock Vina outperformed AutDock in the ability to differentiate actives and decoys. There exist a correlation between AutoDock Vina and AutoDock rankings of active molecules and Demethyloleuropein aglycone was the top-ranked compound. Molecular dynamics analysis predicted that Demethyloleuropein aglycone is a potent non-covalent inhibitor of SARS-CoV-2 PLpro based on its nature and stability of interactions with multiple binding sites. The results reveal the potential of dietary olive secoiridoids in inhibiting PLpro, therefore the Arabian and Mediterranean diets have beneficial effects on host anti-viral defense. Olive secoiridoids are suitable for further preclinical assays and structural optimization studies, leading to better antiviral drugs.
Disclosure of funding
This research was funded by the Deanship of scientific research, Jazan University, Saudi Arabia, Grant No: CoV19-24.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Links between the Prophet Muhammad (PBUH) recommended foods and disease management: A review in the light of modern superfoods. Int. J. Health Sci.. 2018;12:61-69.
- [Google Scholar]
- Dietary adherence of Saudi males to the Saudi dietary guidelines and its relation to cardiovascular diseases: a preliminary cross-sectional study. J. Cardiovasc. Dev. Dis.. 2019;6:17.
- [CrossRef] [Google Scholar]
- Healthy dietary patterns decrease the risk of colorectal cancer in the Mecca Region, Saudi Arabia: a case-control study. BMC Public Health.. 2017;17:1-8.
- [Google Scholar]
- Evaluation and optimization of virtual screening workflows with DEKOIS 2.0–a public library of challenging docking benchmark sets. J. Chem. Inf. Model.. 2013;53:1447-1462.
- [Google Scholar]
- OliveNet™: a comprehensive library of compounds from Olea europaea. Database.. 2018;1–9
- [CrossRef] [Google Scholar]
- Potential protective role exerted by secoiridoids from Olea europaea L. in cancer, cardiovascular, neurodegenerative, aging-related, and immunoinflammatory diseases. Antioxidants.. 2020;9:149.
- [CrossRef] [Google Scholar]
- Secoiridoids of olive and derivatives as potential coadjuvant drugs in cancer: A critical analysis of experimental studies. Pharmacol. Res.. 2019;142:77-86.
- [Google Scholar]
- Virtual screening for HIV protease inhibitors: a comparison of AutoDock 4 and Vina. PloS one.. 2010;5:e11955.
- [Google Scholar]
- Chemical constituents from coconut waste and their in silico evaluation as potential antiviral agents against SARS-CoV-2. S Afr. J. Bot.. 2021;141:278-289.
- [Google Scholar]
- Potential uses of olive oil secoiridoids for the prevention and treatment of cancer: A narrative review of preclinical studies. Int. J. Mol. Sci.. 2021;22:1234.
- [CrossRef] [Google Scholar]
- Screening explorer–An interactive tool for the analysis of screening results. J. Chem. Inf. Model.. 2016;56:2281-2286.
- [Google Scholar]
- Traditional uses, phytochemistry, and pharmacology of Olea europaea (olive) Evid Based Complement. Alternat. Med.. 2015;541591
- [CrossRef] [Google Scholar]
- Supporting SARS-CoV-2 papain-like protease drug discovery: In silico methods and benchmarking. Front. Chem.. 2020;996
- [CrossRef] [Google Scholar]
- Health-promoting properties of oleocanthal and oleacein: Two secoiridoids from extra-virgin olive oil. Crit. Rev. Food Sci. Nutr.. 2020;60:2532-2548.
- [Google Scholar]
- Total analysis of the major secoiridoids in extra virgin olive oil: Validation of an UHPLC-ESI-MS/MS method. Antioxidants.. 2021;10:540.
- [CrossRef] [Google Scholar]
- Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des.. 2013;27:221-234.
- [Google Scholar]
- The secrets of the Mediterranean diet. Does [only] olive oil matter? Nutrients. 2019;11:2941.
- [CrossRef] [Google Scholar]
- AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem.. 2009;30:2785-2791.
- [Google Scholar]
- Oleuropein, a bioactive compound from Olea europaea L., as a potential preventive and therapeutic agent in non-communicable diseases. Antioxidants. 2019;8:578.
- [CrossRef] [Google Scholar]
- Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun.. 2021;12:1-9.
- [Google Scholar]
- Combined docking methods and molecular dynamics to identify effective antiviral 2, 5-diaminobenzophenonederivatives against SARS-CoV-2. J. King Saud Univ. Sci.. 2021;33:101352
- [CrossRef] [Google Scholar]
- Decoys selection in benchmarking datasets: overview and perspectives. Front. Pharmacol.. 2018;9:11.
- [CrossRef] [Google Scholar]
- Integrating molecular docking and molecular dynamics simulations. Methods Mol Biol.. 2019;2053:13-34.
- [CrossRef] [Google Scholar]
- Potential role of medicinal plants and their constituents in the mitigation of SARS-CoV-2: identifying related therapeutic targets using network pharmacology and molecular docking analyses. RSC Advances.. 2020;10:27961-27983.
- [Google Scholar]
- Design of SARS-CoV-2 PLpro Inhibitors for COVID-19 Antiviral Therapy Leveraging Binding Cooperativity. J. Med. Chem.. 2022;65:2940-2955.
- [CrossRef] [Google Scholar]
- Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature.. 2020;587:657-662.
- [Google Scholar]
- AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem.. 2010;31:455-461.
- [Google Scholar]
- End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem. Rev.. 2019;119:9478-9508.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2022.102402.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




