

Benchmark taxonomic classification of chicken gut bacteria based on 16S rRNA gene profiling in correlation with various feeding strategies
⁎Corresponding authors. sadia.galani@kibge.edu.pk (Saddia Galani)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Taxonomic characterization of bacterial 16S rRNA gene has become an accepted norm for inference of complex microbial communities and ecological shifts in gut. Recently, 16S rRNA gene sequences has emerged as a taxonomic marker to investigate crosstalk between gut bacteria and host via variations in feed. Here, broiler chickens (n = 240) were subjected to dietary modulation approach to substitute antibiotics with natural plant-derived products. Five experimental groups were control, antibiotic growth promoter, organic acids, phytogenic feed additives and combination (organic acids + phytogenic feed additives). Bacterial DNA was extracted and 16S rRNA gene was amplified through polymerase chain reaction using universal primers followed by observation through gel electrophoresis and then, confirmation by direct DNA sequencing. The sequence data was analyzed for homology search, multiple sequence alignment, and construction of phylogenetic clades using software BLASTn, CLUSTAL W and MEGA 7.0. respectively. Based on the phenetic relationship, bacterial phylotypes from organic acid and phytogenic feed additive groups belongs to Enterococcus faecium and Enterococcus faecalis in both cecum and ileum. The bacterial isolate obtained from antibiotic feeding group characterized as Escherichia coli. Furthermore, sequences of probiotic bacteria (Enterococcus faecalis) were retrieved from online repositories (GenBank and RDP) and aligned with indigenously isolated strains of this study. Comparative phylogenetic analysis revealed high sequence similarities (99%) of Enterococcus faecalis KIBGE AB-6 and Enterococcus faecalis KIBGE AB5 with Enterococcus faecalis ATCC 19433, and Enterococcus faecalis DD28 respectively. Hence, under the appropriate conditions of current investigation, these bacterial strains might serve as potential candidates for probiotics in animals. To the best of our knowledge, this is the first study in Pakistan depicting taxonomic classification of chicken gut bacterial diversity from cecal and ileal samples linked with dietary modulation.
Keywords
16S rRNA gene
Broiler chicken
Gut microbiota
Probiotic bacteria
Taxonomy

1 Introduction
Bacterial 16S rRNA gene is a gold standard taxonomic marker used for bacterial species identification and characterization, be it from single isolate or environmental samples (Rivoire and Leibler, 2014). Variation in hereditary material, common choices, and adaptive behaviors broadens the grouping of organisms into different kinds based on speciation occasion process (Shcherbakov, 2010; Sarma et al., 2019). With the initiation of phylogenetic framework for bacterial classification together with the recombinant DNA and sequencing, 16S rRNA gene sequences had sidestep the need of culturing organisms for identification of environmental microbes (Khan et al., 2016). The 16S rRNA gene can easily amplified directly from environmental DNA as PCR product or recombinant DNA clones and subsequently sequenced to identify and genotype environmental microbes. These sequences serve as incisive identifiers for bacteria and thus, facilitate the understanding of correlation between microbial ecosystem and host (Peck et al., 2019; Peters et al., 2018). The sequences of conserved regions in 16S rRNA gene are nearly similar in all bacterial species whereas, presence of hypervariable regions allow species differentiation (Karlsholm, 2017; D’Amore et al., 2016). The accurate inference of 16S rRNA gene sequences as a tool for identification of microbial world rely on two key components: complete unambiguous nucleotide sequences deposition in online databases and assigning the right “label” to each deposited sequence. In addition, purity of isolate, methods of DNA extraction and presence of chimera molecule formations can affect final identification of bacteria (De Vrieze et al., 2018; Fouhy et al., 2016).
The gastrointestinal tract harbors an intricated microbial community which includes yeasts, bacteria, bacteriophages, ciliate protozoans, archaea and anaerobic fungi (Yoon et al., 2017; Morovic et al., 2016). A highly dynamic intestinal microbiota composition exists in chicken gut with spatial shifts in different regions of GI tract linked with the variation in environmental conditions in each compartment (Celi et al., 2017). Bacteria are predominant organisms in gut and outnumber the host cells by ten to one (Torok et al., 2011). After the ban on in-feed antimicrobials by European Union (2006), a substantial interest has generated in exploring gut flora composition in livestock production. Extensive antibiotics usage in poultry industry has linked with the dissemination of infectious agents (Salmonella, Campylobacter, antimicrobial resistance Escherichia coli (ExPEC)) through contaminated eggs and meat. This alarming situation triggered public health concerns to focus on poultry as potential reservoir of antimicrobial resistance (AMR) (Allerberger, 2016).
Biotechnology poses a significantly important impact in poultry nutrition and plays a vital role in feed industry. In this regard, a continuous effort is put forward by nutritionists for production of better and economical feed. However, utilization of feed in host is equally important. For this, various feed additives (enzymes, phytogenic feed additives, prebiotics, probiotics and organic acids) have been used widely in poultry production (Kabir, 2009). These additives has been recognized as potential substitutes to antibiotics for improvement of animal growth and increasing beneficial bacterial population through modification of gut environment and associated bacteria (Ahsan et al., 2018; Attia et al., 2017). Ecological studies on chicken gut bacteria revealed positive relationship between probiotic bacteria and increased host health (Wakita et al., 2018). The currently used probiotic bacterial genera in animal diet are Lactobacillus, Enterococcus and Bifidobacterium. The gut bacterial mode of action mainly includes stabilizing host gut ecosystem through antagonism and competitive exclusion, improving feed intake through increasing digestive enzymes activities and enhancing immunity.
The current study is designed for identification and taxonomic characterization of chicken gut bacteria in cecum and ileum regions using 16S rRNA marker through dietary variations. Also, comparative genetic and phylogenetic analysis of indigenously isolated probiotic bacterial sequences were performed for inference of probiotic strains. These phylogenetic implications will help to substitute in-feed antibiotics to combat cross-contamination of poultry pathogens.
2 Materials and methods
2.1 Ethics statement
This research was approved by The Karachi Institute of Biotechnology and Genetic Engineering (KIBGE), University of Karachi ethical review board (DG/AA-089).
2.2 Chickens, study groups and sampling
Two-hundred-fifty-day-old broiler chicks (hubbard strain) were procured from commercial hatchery. Chicks were weighed and inspected upon receive for any signs of deformity and disease. The study was conducted on healthy chicks. Standard operating procedures of animal housing management (broiler) were followed during the trial according to “Care and use of agricultural animals in research” (McGlone, 2010). Before the experiment, disinfection and cleaning of chicken cages, feed trough, drinkers, and feeders were executed.
Healthy birds were allocated five experimental groups with five replicates having ten birds/replicates. To every replicate a clean cage (250 cm × 250 cm) was assigned with rice hulls as litter and self-feeder and waterer was provided. The provision of lightning was 100 W bulb per cage for 7 days. The chickens fed commercial starter (1–21 days) and finisher (21–42 days) diets ad libitum (Table 1).
| Nutrient composition | Starter diet | Finisher diet |
|---|---|---|
| 0–21 days | 22–42 days | |
| Metabolize Energy (Kcal /kg) | 2900.5 | 2989.3 |
| Calcium (%) | 1.02 | 0.89 |
| Crude Protein (%) | 21.59 | 19.13 |
| Available Phosphorous (%) | 0.45 | 0.39 |
| Dig. Methionine (%) | 0.55 | 0.45 |
| Dig. Lysine (%) | 1.20 | 1.06 |
| Dig. Cysteine + Methionine (%) | 0.90 | 0.74 |
| Dig. Tryptophan (%) | 0.28 | 0.19 |
| Dig. L-Threonine (%) | 0.84 | 0.72 |
Study groups received commercial corn-soybean basal diet prepared with following variations:
A: Control (CON): basal diet (no feed additives)
B: Antibiotic growth promoter (AB): basal diet + enramycin (125 mg/kg of diet)
C: Organic acids (ORG): basal diet + organic acids (Citric, formic, lactic acid, MCFA, mono-, di-&triglycerides MCFA, 2 gm/kg of feed)
D: Phytogenic feed additives (PFA): basal diet + blend of Allium sativa (10%), Mentha piperita (10%), Cinnamomum verum (10%), Cuminum cyminum (10%), Camellia sinensis (10%), 2 gm/kg of feed)
E: Combination (COM): basal diet + combination (organic acids & phytogenic feed additives, 2 gm/kg of feed)
At day 42, two birds were selected randomly from each group for scarification through throat cutting with a knife. Gut samples were scrapped from ileum and cecum region aseptically and immediately stored in −80 °C for future analysis.
2.3 Bacterial culture isolation
Bacterial isolates were cultured on plat count agar plates (Oxoid UK) for isolation of bacterial strains from each treatment. Isolates were grown after serial dilution of 1gm sample (cecal and ileal contents) in phosphate buffer saline and plated on agar media using spread plate technique (for yielding single colonies) followed by aerobic and anaerobic incubation at 37 and 42 °C for 24 h respectively. The bacterial growth colonies were examined on media plates after incubation. Bacterial strains were purified by culturing morphologically different colonies on growth media plates. Initially twenty (20) strains were isolated but among them 10 were completely characterized and sequenced.
2.4 DNA extraction
Bacterial DNA was extracted from pure culture through standard phenol-chloroform method with some modifications (Wilson, 2001). The additional step of RNase A (20 mg/ml) was performed prior to phenol/chloroform/isoamyl (25:24:1) step to remove RNA contamination from the samples. The extraction and purification of genomic DNA was performed, and DNA concentration was quantified on Nanophotometer followed by re-suspension of DNA in tris-EDTA buffer and stored in −20 °C.
2.5 16S rRNA genotyping
The 16S rRNA gene was genotyped through polymerase chain reaction using two sets of primers for amplification. The sequences of primers are: F, 5′-AGAGTTTGATCCTGGCTCAG-3′ (20 bp) and R, 5′-GGTTACCTTGTTACGACTT-3′ (19 bp) covers full length 16S rRNA gene (1541 bp) (Chen et al., 2015). The reaction mixture used was 50 µl encompassing 25 ng/µl of gDNA with 1.5 mM Mg+2, 0.2 mM dNTPs, 0.2 µM primers, and 1U/µl Taq polymerase (MOLEQULEON, Auckland, New Zealand). The PCR reaction conditions included first temperature hold for 5 min at 95 °C, subsequently denaturing gDNA for 1 min at 95 °C. The annealing and elongation of primers were done for 45 s and 1 min at 55 and 72 °C at each step respectively. The complete cycle was repeated for 35 times prior to final extension for 10 min at 72 °C. The amplified product (approximately 1542 bp) was observed on 2.0% agarose gel with DNA staining dye SYBR® Safe under ultraviolet light of gel documentation instrument (Gel Doc 2000, BioRad, USA).
2.6 DNA sequencing of 16S rRNA gene
The bands were excised separately from agarose gel and each band was purified through AccuPrep® PCR purification kit (Bioneer, Korea) as per protocol described in kit. The purified PCR products were sequenced by Bioneer Corporation, Korea.
2.7 Bioinformatics analysis
The amplicon data AB1 files after sequencing was loaded in associated software which generates chromatogram and data in FASTA format. DNA sequences obtained were trimmed manually before loading in online software basic local alignment tool (BLASTn). The sequences were searched for similarity with the already available sequences in GenBank using BLASTn. The sequences with similarity index ≤97% were retained for further analysis. For construction of phylogenetic tree to study taxonomic clustering of bacterial isolates, sequences were subjected to multiple sequence alignment (MSA) using online tool CLUSTAL W. After alignment of sequences, phylogenetic clades were reconstructed employing molecular evolutionary genetic analysis (MEGA 7.0.) software. The reconstructed phylogenetic trees by neighbor-joining method depicted confidence percentages of each operational taxonomic units (OTUs) position within tree. To validate the taxonomic tree, 1000 bootstrap clades were built which resulted in a consensus tree depicting estimated bootstrap value. The bootstrap of each placement was provided with the percentage value on each branch point. Furthermore, sequences of probiotic bacteria Enterococcus faecalis were retrieved from online databases (GenBank and RDP) (Al Atya et al., 2015). The sequences of isolated strains along with the retrieved sequences were aligned and phylogenetic tree was reconstructed estimating genetic similarity of novel potential probiotic strains of current study.
3 Results
3.1 Bacterial strains isolation
In total, ten bacterial isolates were used from five treatment groups for studying bacterial phylogenetic analysis. Two out of ten strains showed low similarity during homology search so, these sequences were not considered for phylogenetic tree reconstruction and thus were not assigned any operational taxonomic unit (OTU)) (Table 2).
| Strain ID | Study Group1 | Medium2 | Isolation Source | Strain ID | Taxonomic Assignment |
|---|---|---|---|---|---|
| Isolate I | CON | PCA | Cecum | KIBGE-AB1 | Enterococcus faecium |
| Isolate II | AB | PCA | Cecum | KIBGE-AB3 | Escherichia coli |
| Isolate III | ORG | PCA | Cecum | KIBGE-AB8 | Escherichia coli |
| Isolate IV | PFA | PCA | Cecum | KIBGE-AB6 | Enterococcus faecalis |
| Isolate V | COM | PCA | Cecum | KIBGE-AB4 | Escherichia coli |
| Isolate VI | CON | PCA | Ileum | Low Similarity | Low Similarity |
| Isolate VII | AB | PCA | Ileum | Low Similarity | Low Similarity |
| Isolate VIII | ORG | PCA | Ileum | KIBGE-AB5 | Enterococcus faecalis |
| Isolate IX | PFA | PCA | Ileum | KIBGE-AB7 | Enterococcus faecalis |
| Isolate X | COM | PCA | Ileum | KIBGE-AB2 | Enterococcus faecium |
Study groups1: CON = Control, AB = Antibiotic growth promoter, ORG = organic acids, PHY = phytogenic feed additives, COM = combination; PCA2 = plate count agar.
3.2 16S rRNA genotypic analysis
For 16S rRNA gene amplification standard PCR was performed on bacterial DNA samples from all five treatment groups, followed by agarose gel electrophoresis. The PCR amplicons were represented as distinct fragments of DNA for each group. A negative control for PCR is used for validation of experiment. The amplicons of ∼1542 bp signifies full length 16S rRNA gene. Fig. 1 represents successful bacterial 16S rRNA gene amplification in chicken gut samples (lane 1–10).
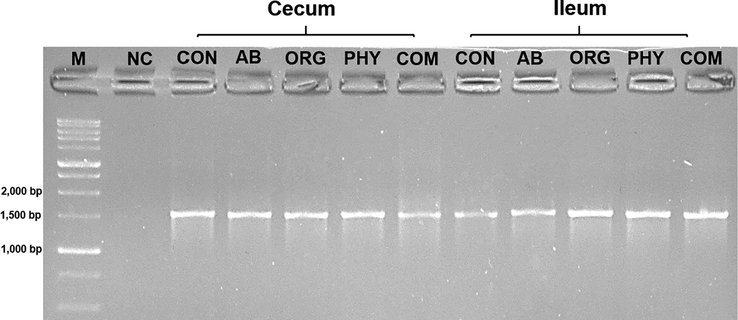
- PCR analysis of full length 16S rRNA gene (∼1542 bp). Lane M: 1 kb DNA ladder. Lane 1–10: Amplified PCR product of bacterial isolates. NC refers to negative/reagent control in the reaction. Study groups: CON = Control, AB = Antibiotic growth promoter, ORG = organic acids, PHY = phytogenic feed additives, COM = combination.
3.3 Phylogenetic analysis of bacterial isolates from study groups
Phylogenetic studies on bacterial isolates from cecum and ileum regions were carried out to infer taxonomic relatedness among bacterial strains inhabiting chicken gut based on dietary modulation. Sequence data were subjected to multiple sequence alignment for reconstruction of phylogenetic trees by Neighbor-joining (NJ) method and substitution model based on maximum composite likelihood with a bootstrap value of 1000.
Phylogenetic tree of each bacterial isolate was created based on minimum evolutionary distance to give a well resolved tree. It was found that isolate I (KIBGE-AB1) was under the group of Enterococcus spp. and closely related to identified organism Enterococcus faecium 7039809-1 with a strong similarity of 99% branch value (Fig. 2). The isolate II (KIBGE-AB3) from AB group is under Enterobacteriaceae family and closely related to Escherichia coli with high similarity index (91%) with already identified organisms (Fig. 3).
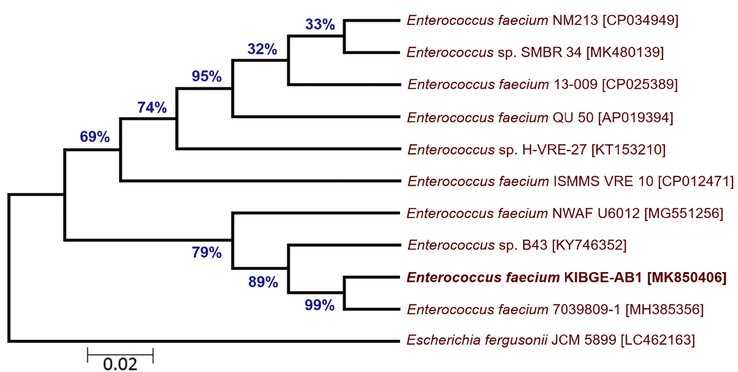
- Phylogenetic relationship of Enterococcus faecium KIBGE-AB1 with related genus and species of Enterococcus.
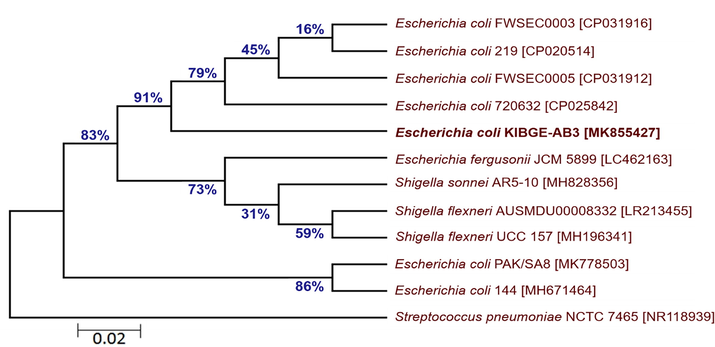
- Phylogenetic relationship of Escherichia coli KIBGE-AB3 with related genus and species of Enterobacteriaceae.
The phylogenetic tree of bacterial isolates from organic acids and combination groups showed that isolate III (KIBGE-AB8) and isolate V (KIBGE-AB4) both are identified as Escherichia coli under the family Enterobacteriaceae. The isolate V (KIBGE-AB4) from combination group is closely related to Escherichia coli BEE25 with 90% similarity index. Against the outgroup Streptococcus pneumoniae, isolate III (KIBGE-AB8) forms distinct branch with 99% similarity to other Escherichia coli strains in the phylogenetic tree. The two indigenously isolated strains KIBGE-AB4 and KIBGE-AB8 from chicken cecum were placed in different branches depicting sequence variations between the two strains (Fig. 4). The isolate IV (KIBGE-AB6) from phytogenic feed additive group was under the group of Enterococcus spp. and closely related to identified organism Enterococcus faecalis CMFRI_FN01 with a strong similarity of 99% branch value (Fig. 5).
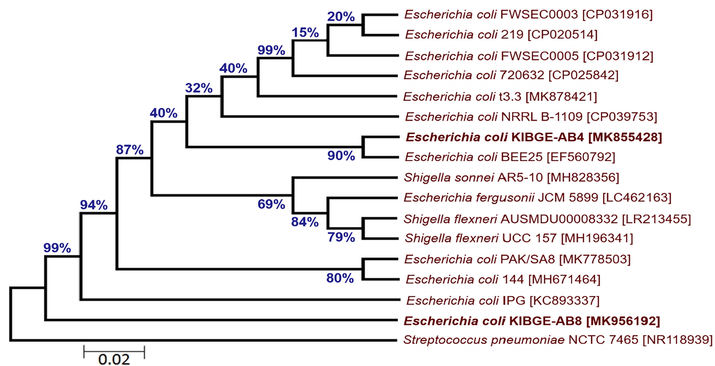
- Phylogenetic relationship of Escherichia coli KIBGE-AB4 and Escherichia coli KIBGE-AB8 with related genus and species of Enterobacteriaceae.
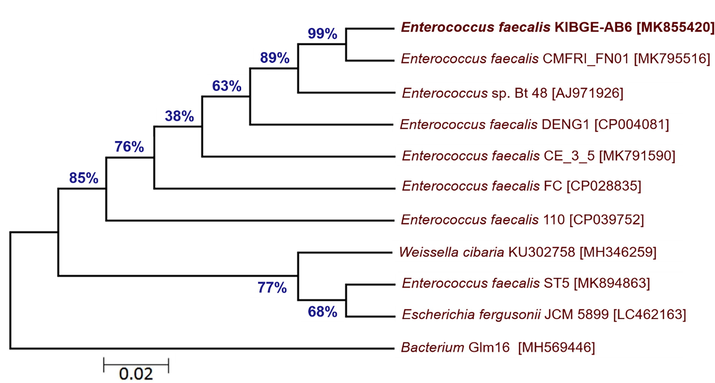
- Phylogenetic relationship of Enterococcus faecalis KIBGE-AB6 with related genus and species of Enterococcus.
From chicken ileum, isolate VI and isolate VII of control and antibiotic growth promoter group were sequenced respectively. The DNA sequences of both the isolates were searched in BLASTn against already submitted sequences. Due to low query coverage these sequences were not considered in phylogenetic tree construction for inference of novel strains. On the basis of maximum likelihood, phylogenetic trees of ileal bacterial isolates from organic acid and phytogenic feed additives groups were reconstructed. The ileal bacterial isolate VIII (KIBGE-AB5) and isolate IX (KIBGE-AB7) were identified as Enterococcus faecalis under the family Enterococcaceae. Fig. 6 shows division of phylogenetic tree in two clades with an outgroup (Streptococcus pneumoniae NCTC 7465). The ileal isolate X (KIBGE-AB2) from combination group was classified as Enterococcus faecium and closely related to Enterococcus faecium E8407 with 87% branch similarity value (Fig. 7).
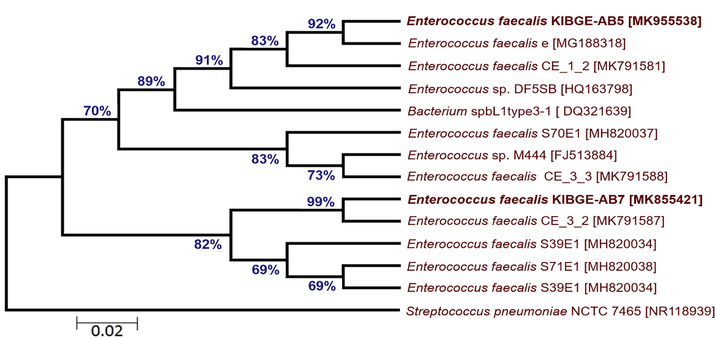
- Phylogenetic relationship of Enterococcus faecalis KIBGE-AB5 and Enterococcus faecalis KIBGE-AB7 with related genus and species of Enterococcus.
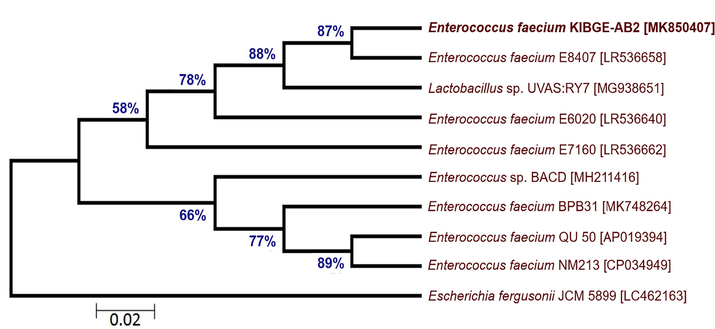
- Phylogenetic relationship of Enterococcus faecium KIBGE-AB2 with related genus and species of Enterococcus.
3.4 Inference of phylogenetic relationships of indigenously isolated probiotic bacteria
The sequences of indigenously isolated Enterococcus faecalis strains from organic acids and phytogenic feed additive (PFAs) groups were phylogenetically characterized based on 16S rRNA gene to decipher their probiotic potential. Sequences were aligned (Fig. 8) with already submitted probiotic bacterial sequences and phylogenetic tree was reconstructed.
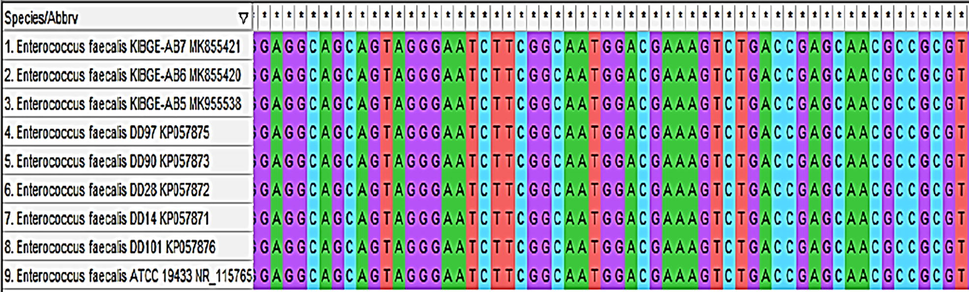
- Sequence alignment of Enterococcus faecalis KIBGE-AB5, Enterococcus faecalis KIBGE-AB6 and Enterococcus faecalis KIBGE-AB7 with probiotic bacterial strains.
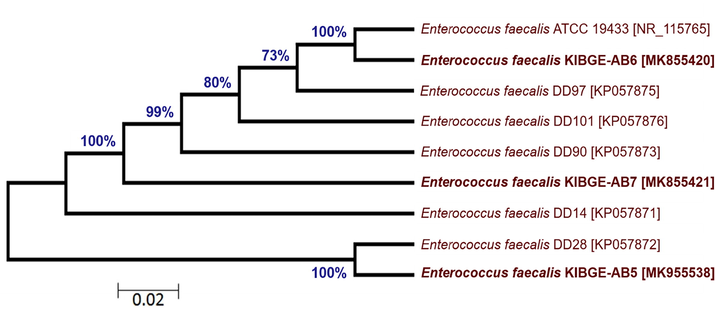
The phylogenetic tree (Fig. 9) depicts high similarity indices of genetic relatedness on the basis of 16S rRNA gene sequences. The highly branched clade revealed 100% similarity between Enterococcus faecalis KIBGE-AB6 and Enterococcus faecalis ATCC 19433. Another isolate Enterococcus faecalis KIBGE-AB5 is also 100% similar to Enterococcus faecalis DD28. The third isolate Enterococcus faecalis KIBGE-AB7 is placed in a separate branch with similarity value of 99% to the different probiotic strains. Notably, the reference sequences used for comparison, alignment and phylogenetic distance estimation are already tested and confirmed as probiotic bacteria (Al Atya et al., 2015).
- Phylogenetic relationship of Enterococcus faecium KIBGE-AB2 with related genus and species of Enterococcus.
3.5 Accession numbers
The bacterial sequences were deposited in GenBank with the accession numbers as MK850406 (Enterococcus faecium KIBGE-AB1), MK850407 (Enterococcus faecium KIBGE-AB2), MK855427 (Escherichia coli KIBGE-AB3), MK855428 (Escherichia coli KIBGE-AB4), MK955538 (Enterococcus faecalis KIBGE-AB5), MK855420 (Enterococcus faecalis KIBGE-AB6), MK855421 (Enterococcus faecalis KIBGE-AB7) and MK956192 (Escherichia coli KIBGE-AB8).
4 Discussion
In the current study, on the basis of 16S rRNA gene variation unknown bacterial cultures were isolated, sequenced and analyzed by bioinformatics tools to decipher their taxonomic assignment. The data from all study groups in cecum and ileum region showed that the bacterial isolates mainly belonged to Enterococcus faecium, Enterococcus faecalis and Escherichia coli.
Recently, gut microbiota emerged as an animal performance biomarker for estimation of animal health. In the light of various challenges (strain selection, use of antibiotics, raised costs, and environmental changes) faced by poultry industry the supply of disease free, healthy, and economically stable poultry is quintessential (Ahsan et al., 2018; Tindall et al., 2010). Phytogenic feed additives (PFA) consist of variety of bioactive compounds which proliferates the growth of beneficial bacteria thus, aiding the host in proper utilization of energy from diet. Similarly, organic acids are also used in chicken feed because of their ability to lower gut pH which is unfavorable for most pathogenic bacteria (Dittoe et al., 2018; Khan and Iqbal, 2016; Suresh et al., 2018). Bacterial cultures isolated from phytogenic feed additive (PFAs) and organic acid groups predominantly belong to E. faecalis and E. faecium. Both are bacteriocin producing strains of Enterococci. Their presence in natural growth promoter groups depicted that these feed additives promote the growth of probiotic bacteria in chicken gut which enhances animal health and efficiency.
The lactic acid bacteria, Enterococci includes a variety of beneficial and pathogenic microorganisms found ubiquitously in environment mostly as gut symbionts. Enterococci have the ability to tolerate change in pH, temperature and salt concentrations which makes them highly adaptive to several food systems (Hanchi et al., 2018). Some strains of Enterococci produce bacteriocins, which is applied for food products preservation and currently being considered as a probiotic trait. Besides, to combat emerging antimicrobial resistance bacteriocins are being recognized as promising alternatives (Gupta and Tiwari, 2015; Hammami et al., 2013; Henning et al., 2015).
Among Enterococci, discrete strains of E. faecalis and E. faecium are being used as probiotics or feed additives because of bacteriocin production. The produced bacteriocin of Enterococci is commonly known as enterocins. The main producers of enterocins in genus Enterococcus are E. faecium and E. faecalis and to a minor extent E. hirae, E. durans and E. avium (Aguayo et al., 2018; Fisher and Phillips, 2009). Enterocins shows anti-microbial activity against both gram-negative and gram-positive bacteria. E. faecium produces enterocin AS-48 which inhibits growth of Listeria and is also active against methicillin-resistant Staphylococcus aureus (MRSA) and prevents film formation. Notably, E. faecium ST5Ha produces bacteriocin ST5Ha which exhibits antiviral activity against Herpes simplex virus HSV1 and HSV2 (Al Atya et al., 2015; Khan et al., 2010; Todorov et al., 2010). The main factor to distinguish between pathogenic Enterococcus and probiotic Enterococcus strains is ampicillin sensitivity. Probiotic Enterococcus strains which can be supplied in animal feed should be susceptible to ampicillin (MIC ≤ 2 mg/L) with the absence of these genetic elements hylEfm, IS16, and esp (Brodmann et al., 2017).
Chicken gut bacteria have historically been studied mainly using classical culture-based methods which rely on highly selectivity in culturing bacteria under specific conditions as a result large proportion of uncultured bacteria remain unidentified. Culture-independent analysis of chicken gut microbiota revealed a great diversity in bacterial population across various sections of gastrointestinal tract. Phylogenetic studies of chicken gut bacteria based on 16S rRNA gene using molecular and biotechnological approaches showed dynamic shifts in taxonomic community of cecal flora from Bacteroides, Firmicutes, and Proteobacteria, to nearly complete Firmicutes at 21 days. It has been reported that cecal population mainly comprises of anaerobes such as: Clostridium and Campylobacter spp. contrastingly, the microbial community in ileum is less diverse and predominantly comprises of Lactobacilli (Choi et al., 2015; Shang et al., 2018a). The composition of chicken ileal gut flora using 16S rDNA primers described the abundance of three major genera of bacteria in ileum including Lactobacilli, Enterococcus, and Butyrate-producing bacteria (Shang et al., 2018b). Overall, the exploration of diversity and succession of stable gut flora depends on dietary composition/treatments, breed, environmental factors, sequencing approach, primers and geographical distribution.
5 Conclusions
The current work illustrates that dietary inclusion of phytogenic feed additives (PFAs) and organic acids successfully modulates gut flora and proliferates growth of probiotic bacteria. The indigenously isolated probiotic bacteria of this study might serve as potential probiotic bacteria for animal feed. By using the strategy of 16S rRNA gene profiling unidentified bacteria can be genotyped and characterized. These findings are potentially significant for better understanding and designing nutritional strategies for deciphering chicken gut bacteria. However, these findings are still considered insufficient and require further validation in future using high-through put sequencing approach for extensive analysis of probiotic bacterial strains along with in-depth investigation of other gut performance biomarkers.
Acknowledgements
All authors show gratitude to the administration of Ideal feeds and experimental units, Super-highway, Karachi for providing animal house facility. “The authors (SM, KAG and FAM) express their sincere appreciation to the Researcher Supporting Project No. RSP-2019/48, King Saud University, Riyadh, Saudi Arabia”.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Immobilization of bacteriocin enterocin AS-48 on different supports. Explor. Microorg. Recent Adv. Appl. Microbiol. 2018:87.
- [Google Scholar]
- Dietary supplementation of different levels of phytogenic feed additive in broiler diets: the dynamics of growth performance, caecal microbiota, and intestinal morphometry. Brazilian J. Poult. Sci.. 2018;20:737-746.
- [Google Scholar]
- Probiotic potential of Enterococcus faecalis strains isolated from meconium. Front. Microbiol.. 2015;6:227.
- [Google Scholar]
- Effect of dietary supplementation with a plant extract blend on the growth performance, lipid profile, immune response and carcass traits of broiler chickens. Int. J. Poult. Sci.. 2017;16:248-256.
- [Google Scholar]
- Safety of novel microbes for human consumption: practical examples of assessment in the European Union. Front. Microbiol.. 2017;8:1725.
- [Google Scholar]
- Gastrointestinal functionality in animal nutrition and health: new opportunities for sustainable animal production. Anim. Feed Sci. Technol.. 2017;234:88-100.
- [Google Scholar]
- Obtaining long 16S rDNA sequences using multiple primers and its application on dioxin-containing samples. BMC Bioinformatics. 2015;16:S13.
- [Google Scholar]
- Metagenomic analysis of chicken gut microbiota for improving metabolism and health of chickens—a review. Asian-Australasian J. Anim. Sci.. 2015;28:1217.
- [Google Scholar]
- A comprehensive benchmarking study of protocols and sequencing platforms for 16S rRNA community profiling. BMC Genomics. 2016;17:55.
- [Google Scholar]
- The active microbial community more accurately reflects the anaerobic digestion process: 16S rRNA (gene) sequencing as a predictive tool. Microbiome. 2018;6:63.
- [Google Scholar]
- Organic acids and potential for modifying the avian gastrointestinal tract and reducing pathogens and disease. Front. Vet. Sci. 2018:5.
- [Google Scholar]
- The ecology, epidemiology and virulence of Enterococcus. Microbiology. 2009;155:1749-1757.
- [Google Scholar]
- 16S rRNA gene sequencing of mock microbial populations-impact of DNA extraction method, primer choice and sequencing platform. BMC Microbiol.. 2016;16:123.
- [Google Scholar]
- Probiotic potential of bacteriocin-producing Enterococcus hirae strain LD3 isolated from dosa batter. Ann. Microbiol.. 2015;65:2333-2342.
- [Google Scholar]
- Anti-infective properties of bacteriocins: an update. Cell. Mol. life Sci.. 2013;70:2947-2967.
- [Google Scholar]
- The genus Enterococcus: Between probiotic potential and safety concerns—an update. Front. Microbiol. 2018:9.
- [Google Scholar]
- Identification of multiple bacteriocins in Enterococcus spp. using an Enterococcus-specific bacteriocin PCR array. Microorganisms. 2015;3:1-16.
- [Google Scholar]
- The role of probiotics in the poultry industry. Int. J. Mol. Sci.. 2009;10:3531-3546.
- [Google Scholar]
- Karlsholm, L.S., 2017. Intragenomic variation among 16S rRNA copies in Vibrio-significance of lifestyle. NTNU.
- Role of recombinant DNA technology to improve life. Int. J. Genomics. 2016;2016:1-14.
- [Google Scholar]
- Recent advances in the role of organic acids in poultry nutrition. J. Appl. Anim. Res.. 2016;44:359-369.
- [Google Scholar]
- McGlone, J., 2010. Guide for the care and use of agricultural animals in research and teaching. Federation of Animal Science Societies.
- Genotyping by PCR and high-throughput sequencing of commercial probiotic products reveals composition biases. Front. Microbiol.. 2016;7:1747.
- [Google Scholar]
- Peck, K., Stedtfeld, R., RoseFigura, J., Reed, B., McUsic, D., Irish, J., Etchebarne, B., Johnson, T., Kurihara, L., Makarov, V., 2019. Microbial sequencing using a single-pool target enrichment of multiple variable regions of the 16S rRNA gene, the nuclear ribosomal internal transcribed spacer (ITS) region, and antimicrobial resistance genes.
- 26. Peters, L., Spatharis, S., Hanssen, M.K., Kanstadd, O., JT Roca, I., Kintner, A., Llewellyn, M.S., Praebel, K., 2018. Environmental DNA: a new low-cost monitoring tool for pathogens in salmonid aquaculture. Front. Microbiol. 9, 3009.
- A model for the generation and transmission of variations in evolution. Proc. Natl. Acad. Sci.. 2014;111:E1940-E1949.
- [Google Scholar]
- Sarma, H., Pradhan, S., Mattaparthi, V.S.K., Kaushik, S., 2019. Phylogenetic Analysis: Early Evolution of Life.
- Chicken gut microbiota: Importance and detection technology. Front. Vet. Sci.. 2018;5:254.
- [Google Scholar]
- Effect of dietary fructooligosaccharide (FOS) supplementation on ileal microbiota in broiler chickens. Poult. Sci.. 2018;97:3622-3634.
- [Google Scholar]
- Biological species is the only possible form of existence for higher organisms: the evolutionary meaning of sexual reproduction. Biol. Direct. 2010;5:14.
- [Google Scholar]
- Alternatives to antibiotics in poultry feed: molecular perspectives. Crit. Rev. Microbiol.. 2018;44:318-335.
- [Google Scholar]
- Notes on the characterization of prokaryote strains for taxonomic purposes. Int. J. Syst. Evol. Microbiol.. 2010;60:249-266.
- [Google Scholar]
- Characterisation of an antiviral pediocin-like bacteriocin produced by Enterococcus faecium. Food Microbiol.. 2010;27:869-879.
- [Google Scholar]
- Identification and characterization of potential performance-related gut microbiotas in broiler chickens across various feeding trials. Appl. Environ. Microbiol.. 2011;77:5868-5878.
- [Google Scholar]
- Taxonomic classification for microbiome analysis, which correlates well with the metabolite milieu of the gut. BMC Microbiol.. 2018;18:188.
- [Google Scholar]
- Wilson, K., 2001. Preparation of genomic DNA from bacteria. Curr. Protoc. Mol. Biol. 56, 2.4. 1-2.4. 5.
- Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol.. 2017;67:1613.
- [Google Scholar]




