

Translate this page into:
Artesunate induces substantial topological alterations in the SARS-CoV-2 Nsp1 protein structure
⁎Corresponding author. arunbgurung@gmail.com (Arun Bahadur Gurung)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The need for novel antiviral treatments for coronavirus disease 2019 (COVID-19) continues with the widespread infections and fatalities throughout the world. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of the deadly disease, relies on the non-structural protein Nsp1 for multiplication within the host cells and disarms the host immune defences by various mechanisms. Herein, we investigated the potential of artemisinin and its derivatives as possible inhibitors of SARS-CoV-2 Nsp1 through various computational approaches. Molecular docking results show that artemisinin (CID68827) binds to Nsp1 with a binding energy of −6.53 kcal/mol and an inhibition constant of 16.43 µM. The top 3 derivatives Artesunate (CID6917864), Artemiside (CID53323323) and Artemisone (CID11531457) show binding energies of −7.92 kcal/mol, −7.46 kcal/mol and −7.36 kcal/mol respectively. Hydrophobic interactions and hydrogen bonding with Val10, Arg11, and Gln50 helped to stabilize the protein–ligand complexes. The pharmacokinetic properties of these molecules show acceptable properties. The geometric parameters derived from large-scale MD simulation studies provided insights into the changes in the structural topology of Nsp1 upon binding of Artesunate. Thus, the findings of our research highlight the importance of artemisinin and its derivatives in the development of drugs to inhibit SARS-CoV-2 Nsp1 protein.
Keywords
Artemisinin
Artemisinin derivatives
Artesunate
COVID-19
Nsp1
SARS-CoV-2

1 Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a betacoronavirus that has enveloped, positive-sense, single-stranded genomic RNA of about 29.9 kb and is the cause of the COVID-19 pandemic (Gorbalenya et al., 2020). SARS-CoV-2 has a significant sequence similarity and infection mechanism to other coronaviruses including the SARS-CoV outbreak in 2002, the Middle East Respiratory Syndrome (MERS) epidemic in 2012 and, infecting the lower respiratory tract of the host (Wang et al., 2020). However, the COVID19 epidemic was deemed to be more dangerous than previous coronavirus outbreaks, with greater morbidity and death (Vankadari et al., 2020). It has spread fast throughout the world, causing significant social and economic damage (Min et al., 2020). The SARS-CoV-2 genome codes for two large overlapping open reading frames (ORF1a and ORF1b) in gene 1 as well as several structural and nonstructural accessory proteins (Zhou et al., 2020). SARS-CoV-2 hijacks the translation machinery of the infected cell to produce ORF1a and ORF1b polyproteins, which is then proteolytically cleaved into sixteen mature non-structural proteins, namely Nsp1 through Nsp16 (Hartenian et al., 2020). The N-terminal nonstructural protein 1 (Nsp1) is one of these proteins.
Nsp1 proteins from alpha- and beta-CoVs have similar biological functions in inhibiting host gene expression despite variations in protein size and mechanism of action (Huang et al., 2011a; Narayanan et al., 2008; Tohya et al., 2009). SARS-CoV Nsp1 uses a two-pronged approach to cause a near-complete shutdown of host protein translation: first, it binds the small ribosomal subunit and delays conventional mRNA translation at various points during initiation (Kamitani et al., 2009; Lokugamage et al., 2012). Second, Nsp1 attachment to the ribosome results in endonucleolytic cleavage of host mRNAs and subsequent degradation (Huang et al., 2011b). Interestingly, viral RNA is resistant to this cleavage due to a unique structural interaction between Nsp1 and viral RNA in the 5′ untranslated (UTR) region. (Thoms et al., 2020). Nsp1 suppresses all cellular antiviral defence mechanisms that rely on host factor expression, including the interferon response. This suppression of critical components of the innate immune system may aid virus multiplication and immune evasion (Narayanan et al., 2008; Wathelet et al., 2007). SARS-CoV Nsp1 is a promising therapeutic target because of its important function in suppressing the antiviral immune response (Jauregui et al., 2013; Wu et al., 2020). SARS-CoV-2 Nsp1 shares 84 percent of its amino acid sequence with SARS-CoV, implying comparable characteristics and biological activities (Thoms et al., 2020). There is a high level of structural conservation between SARS-CoV-2 Nsp1 and SARS-CoV Nsp1, although there are certain unique structural characteristics in SARS-CoV-2 Nsp1 that likely contribute to the enhanced stability of the β-barrel fold (Semper et al., 2021). The SARS-CoV C-terminal residues Lys164 and His165 are conserved in beta-CoVs and are required for 40S small ribosomal subunit interaction (Kamitani et al., 2009). The SARS-CoV-2 Nsp1 is a critical virulence factor that facilitates viral replication and inhibits host gene expression and immunological defence (Kamitani et al., 2009; Tanaka et al., 2012).
Because of the rapid infection, as well as the emergence of the second wave of COVID-19 around the world, we need to have a deeper understanding of the disease pathogenesis at the molecular levels to develop better therapeutics. The current treatment for COVID19 includes broad-spectrum antiviral drugs such as Remdesivir (which inhibits RNA-dependent RNA polymerase), Arbidol (which inhibits spike proteins), Ritonavir, Hydroxychloroquine, and a cocktail of other therapeutics (Amawi et al., 2020; Vankadari, 2020). While studies have shown the significance of Nsp1 as a key virulence factor in the pathogenesis of SARS-CoV-2 infection and as a therapeutic drug target, no specific inhibitors of this enzyme have been reported to date. Thus, using a molecular docking and molecular dynamics method, the current study investigates the potential of artemisinin and its derivatives to inhibit the activity of SARS-CoV-2 Nsp1.
2 Materials and methods
2.1 Retrieval of the experimental structure of SARS-CoV-2 Nsp1
The experimental structure of SARS-CoV-2 Nsp1 was retrieved from the Protein Data bank (http://www.rcsb.org/) using accession ID: 7K3N, which is a structure of the globular N-terminal domain of SARS-CoV-2 Nsp1 solved through the X-ray diffraction method at a high resolution of 1.65 Å (Semper et al., 2021). The missing segment between Asp62 and Gly73 were added using the loop modelling method using the MODELLER program (Šali and Blundell, 1993).
2.2 Retrieval of ligands
The three-dimensional structures of artemisinin (CID68827) and its nine derivatives- Dihydroartemisinin or Artenimol (CID6918483), Artemiside (CID53323323), Artemotil (CID3000469), Artemisitene (CID10423777), Beta-Artemether (CID68911), Artemisone (CID11531457), Artesunate (CID6917864), Deoxyartemisinin (CID12814879) along with the reference drug-Remdesivir (CID121304016) were downloaded from PubChem database (Kim et al., 2016).
2.3 Molecular docking
Molecular docking approaches have broadly been used to know the binding modes of ligand and receptor and is widely used in drug discovery. We have used a high-resolution crystal structure of the globular N-terminal domain of Nsp1 from SARS-CoV-2 solved at 1.65 Å (PDB ID: 7K3N). The energy minimized 3 D structures were then converted into docking input file format (i.e. PDBQT). We have used Autodock (version 4.2), which is an extensively used automated tool for protein–ligand docking (Morris et al., 2009). The 3 D-grid box was set to be with dimensions 95 × 95 × 95 Å in XYZ axes centred at x:1.715, y:12.4846, z:47.3237, with a grid point spacing of 0.375 Å. The binding poses were generated by the Lamarckian Genetic Algorithm (Morris et al., 1998). Fifty binding poses were generated for each ligand. After docking, the three-dimensional receptor-ligand complexes were constructed using PyMOL (DeLano, 2002) and the molecular interactions between the protein and compound were evaluated using the LigPlot+ tool version 1.4.5 (Laskowski and Swindells, 2011).
2.4 Pharmacokinetics properties of the compounds
The pharmacokinetic properties of the selected compounds were computed using Swiss ADME program (Daina et al., 2017).
2.5 Molecular dynamics simulations
GROningen MAchine for Chemical Simulations (GROMACS) 2019.2 software package (Van Der Spoel et al., 2005) was used to perform MD simulations on unbound Nsp1 and Nsp1-ligand complexes. The GROMOS96 43a1 force field was used to create the protein's topology. Since the GROMACS software program lacks adequate force field values for small molecules, the PRODRG server (Schüttelkopf and Van Aalten, 2004) was used to generate molecular topologies and coordinate files. The systems were solvated in a cubic box using a simple point charge (SPC) model. 0.15 M counter ions (Na+ and Cl-) were introduced to the solution to neutralize it. The steepest descent approach was used to minimize the energy of all neutralized systems. The equilibration phase was performed independently for 300 ps in NVT (constant volume) and NPT (constant pressure). All systems were kept at 300 K using Berendsen's weak coupling technique (Berendsen et al., 1987), and pressure was kept at 1 bar using a Parrinello-Rahman barostat (Parrinello and Rahman, 1980). The hydrogen atoms were confined at their equilibrium distances and periodic boundary conditions using the SHAKE algorithm. The van der Waals and coulombic interactions cutoffs were also set at 1.0 nm. The LINC method was employed to restrict the bonds and angles. Production runs were executed for 100 ns with time integration using the NPT ensemble. MD analysis was performed using as gmx rms, gmx rmsf, gmx gyrate, gmx sasa, and gmx hbond programs of GROMACS to generate plots of root mean square deviations (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), solvent accessible surface area (SASA), and number of hydrogen bonds (NHBs).
3 Results
The structures of Artemisinin and its nine derivatives used for virtual screening are shown in Fig. 1. The binding poses, binding energies and inhibition constants of the compounds with Nsp1 were determined using molecular docking studies (Table 1). Remdesivir was used as a reference drug for comparing the binding energies of the selected molecules. The reference drug binds to Nsp1 with a binding energy of −4.32 kcal/mol and an inhibition constant of 680.67 µM. The molecular interaction involves five hydrogen bonds- a 3.18 Å hydrogen bond between N4 and OE2 atom of Glu78, a 2.64 Å hydrogen bond between N3 and OH atom of Tyr55, a 2.38 Å hydrogen bond between O2 and OH atom of Tyr55, a 2.64 Å hydrogen bond between O8 and O atom of Pro49 and a 2.84 Å hydrogen bond between O5 and N atom of Gln53 and hydrophobic interactions via Arg16, Leu48, Gln50, Leu51, Glu52, Glu80 and Gly81 (Fig. 2E). The molecular docking results show that Artemisinin (CID68827) binds to Nsp1 with binding energies of −6.53 kcal/mol with an inhibition constant of 16.43 µM. The interaction involves a 2.62 Å hydrogen bond between O1 and backbone nitrogen atom of Ser4 and hydrophobic interactions through residues Leu3, Leu5, Pro6, Leu33, Gly36 and Cys38 (Fig. 2D). The first top-ranked molecule Artesunate (CID6917864) binds to Nsp1 with a binding energy of −7.92 kcal/mol and inhibition constant of 1.57 µM which involves five hydrogen bonds-a 2.86 Å hydrogen bond between O7 and NE2 atom of Gln50, a 3.07 Å hydrogen bond between O8 and NE atom of Arg11, a 3.12 Å hydrogen bond between O6 and N atom of Arg11, a 2.78 Å hydrogen bond between O5 and N atom of Val10 and a 2.99 Å hydrogen bond between O3 and NE2 atom of Gln9 and hydrophobic interactions through Leu8, Val41, Glu42, Glu44 and Val103 (Fig. 2A). The second top-ranked molecule Artemiside (CID53323323) binds to Nsp1 with binding energy of −7.46 kcal/mol and an inhibition constant of 3.42 µM. The molecular interaction involves two hydrogen bonds- one 3.02 Å hydrogen bond between O2 and N atom of Val10 and a 3.29 Å hydrogen bond between O3 and N atom of Arg11 and hydrophobic interactions through Val7, Leu8, Gln9, Glu42, Glu44, Lys45, Val47, Gln50 and Val103 (Fig. 2B). The third high ranked lead Artemisone (CID11531457) binds to Nsp1 with a binding energy of −7.36 kcal/mol and an inhibition constant of 4.06 µM. The molecular interaction involves two hydrogen bonds-a 2.71 Å hydrogen bond between O4 and NE2 atom of Gln50 and a 3.09 Å hydrogen bond between O3 and NE atom of Arg11 and hydrophobic interactions through Leu8, Gln9, Val10, Val41, Lys45, Gly46 and Val47 (Fig. 2C).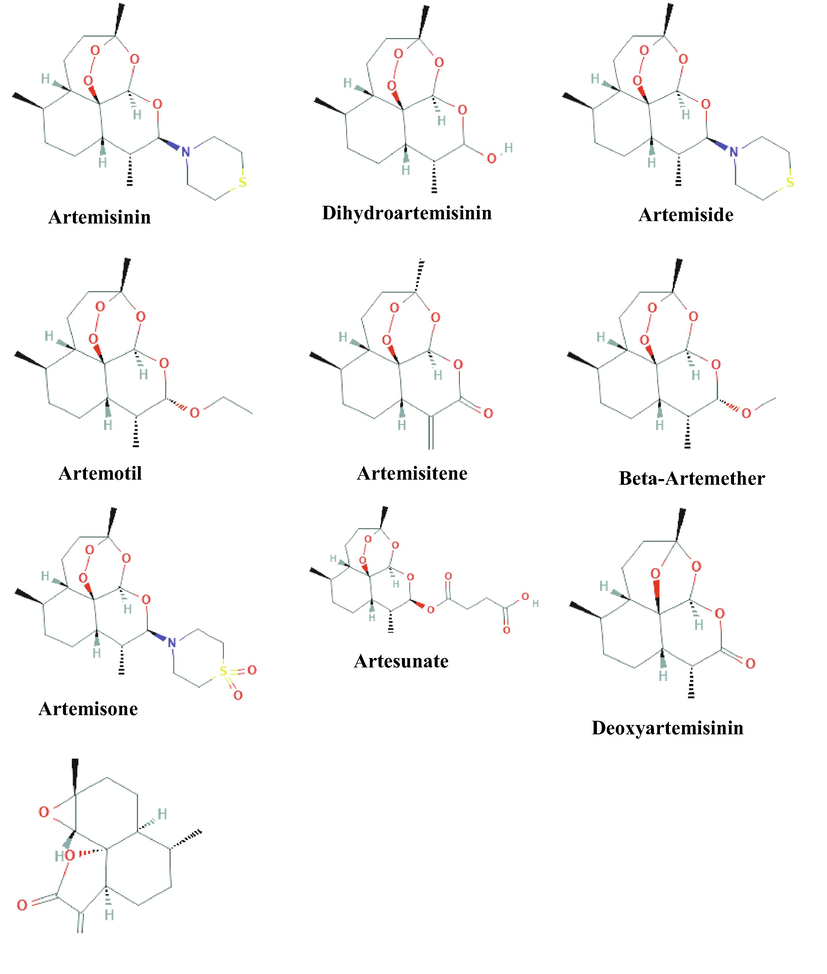
Structures of Artemisinin and its nine derivatives selected for molecular docking studies.
Name
PubChem ID
Molecular formula
Binding Energy (kcal/mol)
Inhibition constant (µM)
Artemisinin
CID68827
C15H22O5
−6.53
16.43
Dihydroartemisinin (Artenimol)
CID6918483
C15H24O5
−6.47
18.22
Artemiside
CID53323323
C19H31NO4S
−7.46
3.42
Artemotil
CID3000469
C17H28O5
−6.72
11.86
Artemisitene
CID10423777
C15H20O5
−6.62
14.16
Beta-Artemether
CID68911
C16H26O5
−6.58
14.95
Artemisone
CID11531457
C19H31NO6S
−7.36
4.06
Artesunate
CID6917864
C19H28O8
−7.92
1.57
Deoxyartemisinin
CID12814879
C15H22O4
−6.26
25.7
Arteannuin B
CID6543478
C15H20O3
−6.45
18.78
Reference drug (Remdesivir)
CID121304016
C27H35N6O8P
−4.32
680.67
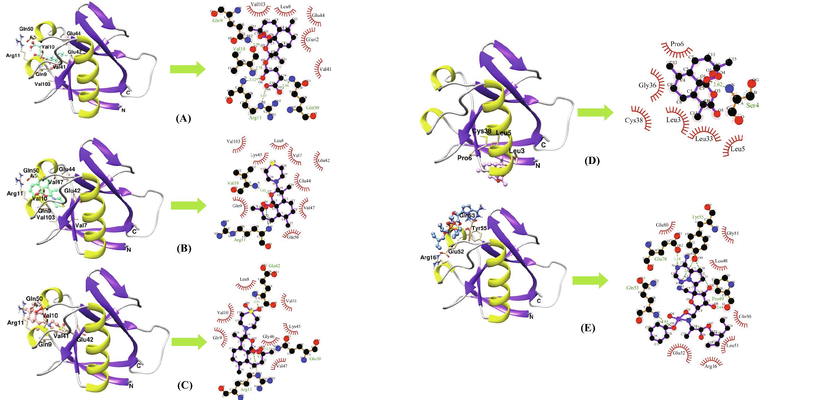
Binding poses and molecular interactions between SARS-CoV-2 and compounds (A) Artesunate (CID6917864) (B) Artemiside (CID53323323) (C) Artemisone (CID11531457) (D) Artemisinin (CID68827) and (E) Remdesivir (CID121304016). Green dashed lines represent hydrogen bonds, whereas red arcs represent hydrophobic interactions. Ligand residues are identified in blue and key protein residues are designated in black (hydrophobic) or red (hydrogen bonds).
The Absorption, Distribution, Metabolism, and Excretion (ADME) characteristics of the compounds were evaluated using the Swiss ADME program. The radar charts which represent bioavailability and the drug-likeness of the compounds contain information such as molecular weight (SIZE), polarity (POLAR), lipophilicity (LIPO), solubility (INSOLU), saturation (INSATU), and flexibility (FLEX) which are within the optimal range (Fig. 3). The data summarized in Table 2 includes information such as solubility, gastrointestinal (GI) absorption, blood–brain barrier (BBB) permeation, P-glycoprotein (P-gp) substrate, cytochrome P450 enzyme inhibitory capacity (subtypes 1A2, 2C9, 2C19, 2D6, 3A4), and skin permeation (log Kp). All the molecules obey Lipinski’s rule of five (i.e. a molecule having a molecular mass of less than 500 Da, no more than 5 hydrogen bond donors, no more than 10 hydrogen bond acceptors, and a log P octanol–water partition coefficient of less than 5) and Veber’s rule (number of rotatable bonds no more than 10 and polar surface area less than 140 Å2) and have acceptable pharmacokinetic properties.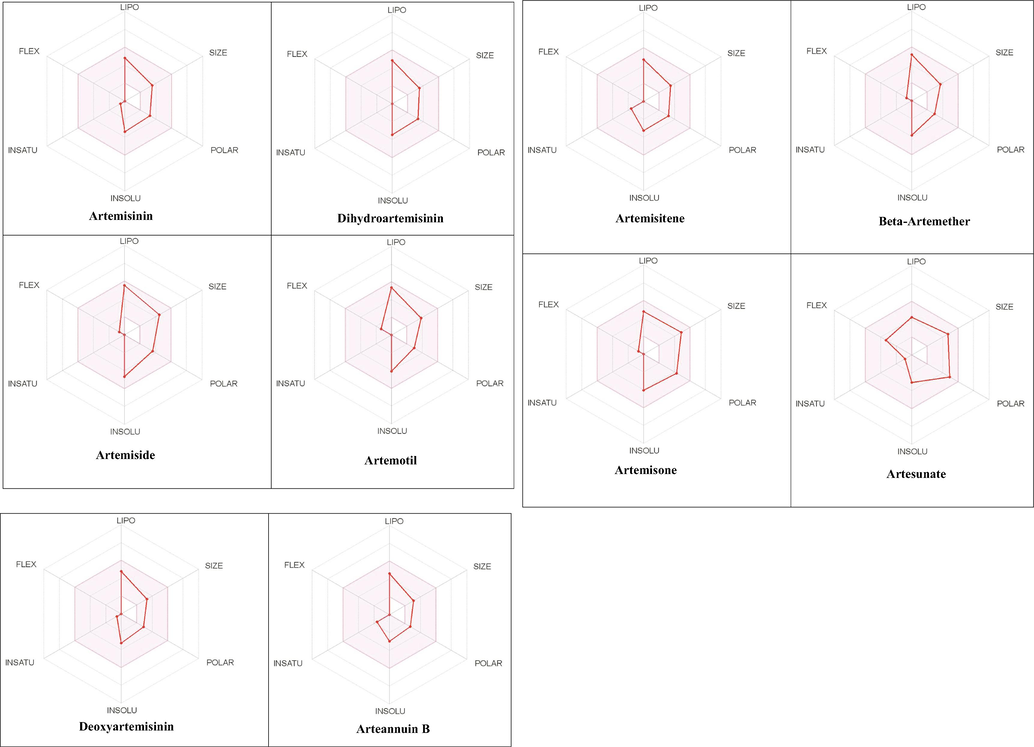
The radar charts depicting bioavailability of compounds. The pink region indicates the ideal range for each properties (lipophilicity: −0.7 < XLOGP3 < +5.0, size: 150 < MW < 500 g/mol, polarity: 20 < TPSA < 130 Å2, solubility: logS < 6, saturation: fraction of sp3 hybridized carbon atoms >0.25, and flexibility: rotatable bonds <9).
Molecule
GI absorption
BBB permeant
Pgp substrate
CYP1A2 inhibitor
CYP2C19 inhibitor
CYP2C9 inhibitor
CYP2D6 inhibitor
CYP3A4 inhibitor
Lipinski #violations
Veber #violations
Bioavailability Score
Leadlikeness #violations
Synthetic Accessibility
Artemisinin
High
Yes
No
Yes
No
No
No
No
0
0
0.55
0
6.13
Dihydroartemisinin
High
Yes
No
No
No
No
No
No
0
0
0.55
0
6.59
Artemiside
High
Yes
No
Yes
No
No
No
No
0
0
0.55
2
6.71
Artemotil
High
Yes
No
Yes
No
No
No
No
0
0
0.55
1
6.76
Artemisitene
High
Yes
No
Yes
No
No
No
No
0
0
0.55
0
5.97
Beta-Artemether
High
Yes
No
Yes
No
No
No
No
0
0
0.55
1
6.65
Artemisone
High
No
No
No
No
No
No
No
0
0
0.55
1
6.8
Artesunate
High
No
No
No
No
No
No
No
0
0
0.56
1
6.67
Deoxyartemisinin
High
Yes
No
Yes
No
No
No
No
0
0
0.55
0
5.48
Arteannuin B
High
Yes
No
No
No
No
No
No
0
0
0.55
1
4.77
A 100-ns all-atom molecular dynamics simulation in an aqueous environment were performed on the Nsp1 protein and its complex with Artesunate (CID6917864) to understand the structural changes in the Nsp1 protein upon binding of the compound. Various geometric properties such as RMSD, RMSF, Rg, SASA and NHB were computed by analysing the MD trajectories (Table 3). To evaluate variations in the backbone atom positions of the Nsp1 protein upon binding of Artesunate, RMSD values were calculated throughout the MD simulations (Fig. 4). Both the Nsp1 and Nsp1_Artesunate show significant deviations in their backbone atoms compared to the starting structures whose average values were found to be 0.250835 ± 0.044315 nm and 0.189268 ± 0.035051 nm respectively (Table 3). The divergence of the backbone atoms of Nsp1_Artesunate with respect to unbound Nsp1 is quite evident within 10–70 ns which sufficiently indicates that the structural topology of nsp1 was significantly altered upon binding of the compound. The compound also seems to be conformationally stable within the binding pocket indicated by a low RMSD value of 1.187178 ± 0.454534 nm (Table 3). The root mean square fluctuation (RMSF) values for backbone atoms at each time point of trajectories of the Nsp1 and Nsp1 artesunate complex were computed to see how the binding of artesunate affects the dynamics of the backbone atoms (Fig. 5). Greater flexibility during the course of MD simulation is shown by higher RMSF values. Except for residues Lys34-Gly36, Ala63-Ala66 and His68, the RMSF values of the Nsp1_artesunate complex are greater than those of the unbound Nsp1. The radius of gyration is a measure of the compactness of the protein and assess the overall conformation changes in the protein over time (Fig. 6). The Nsp1_artesunate complex (1.324615 ± 0.010709 nm) shows higher Rg values compared to the unbound state (1.304486 ± 0.010379 nm) (Table 3). This indicates that the Nsp1 protein has experienced significant structural changes, with a decrease in compactness, as a result of the binding of the compound. Fig. 7 depicts the evolution of total SASA for the Nsp1 and Nsp1_Artesunate complex throughout the simulation period. The bimolecular surface area that may be assessed by solvent molecules is referred to as solvent accessibility surface area. When compared to the unbound Nsp1 structure (64.36101 ± 1.707921 nm2), the increase in SASA value in the Nsp1_Artesunate complex (66.48523 ± 1.623862 nm2) reflects increase surface area exposed to water molecules (Table 3). Non-covalent interactions such as hydrogen bonds contribute to the maintenance of the overall protein structure. The number of intramolecular hydrogen bonds did not change significantly over time (Fig. 8A), although the average number of hydrogen bonds in the Nsp1 artesunate complex (N = 68.42857 ± 4.611198) were higher than in unbound Nsp1 (N = 66.93506 ± 4.851884) (Table 3). The hydrogen bonds between the protein and the compound are one of the molecular forces which strengthen the interactions and stability of the complexes (Fig. 8B). The number of hydrogen bonds between Artesunate and Nsp1 is averaged at 1.178821 ± 0.918145 which allows artesunate to modulate Nsp1 activity (Table 3).
Properties
Nsp1
Nsp1_Artesunate
Protein Backbone RMSD (nm)
0.250835 ± 0.044315
0.189268 ± 0.035051
Ligand RMSD (nm)
–
1.187178 ± 0.454534
Rg (nm)
1.304486 ± 0.010379
1.324615 ± 0.010709
Total SASA (nm2)
64.36101 ± 1.707921
66.48523 ± 1.623862
Intra-protein hydrogen bonds
66.93506 ± 4.851884
68.42857 ± 4.611198
Protein-ligand hydrogen bonds
–
1.178821 ± 0.918145
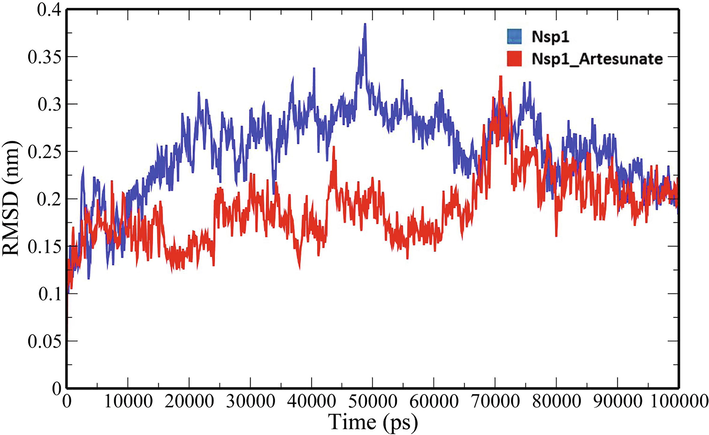
RMSD analysis of backbone atoms of unbound Nsp1 and Nsp1_Artesunate.
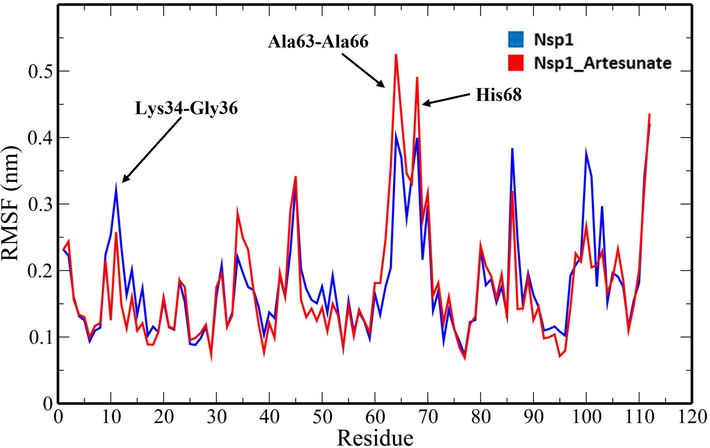
RMSF plot of unbound Nsp1 and Nsp1_Artesunate showing altered drift at the residue level.
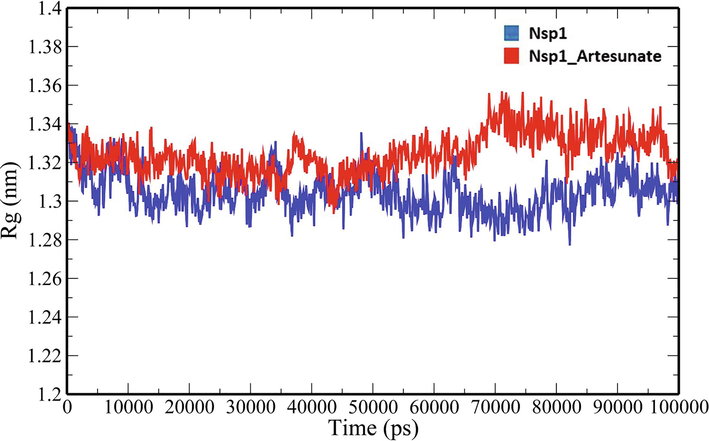
Rg analysis of unbound Nsp1 and Nsp1_Artesunate.
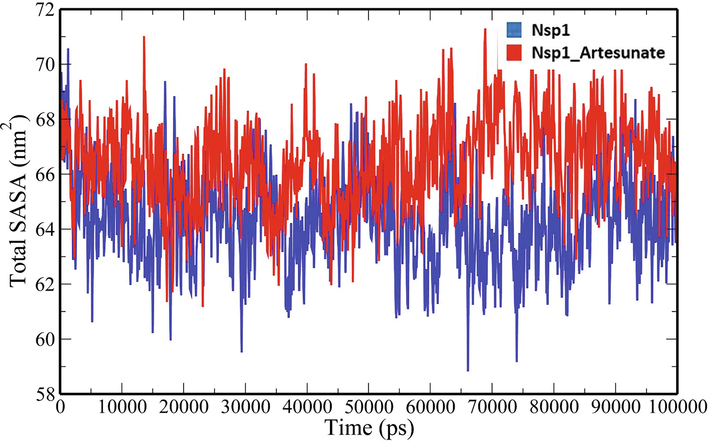
Total SASA analysis of unbound Nsp1 and Nsp1_Artesunate complex.
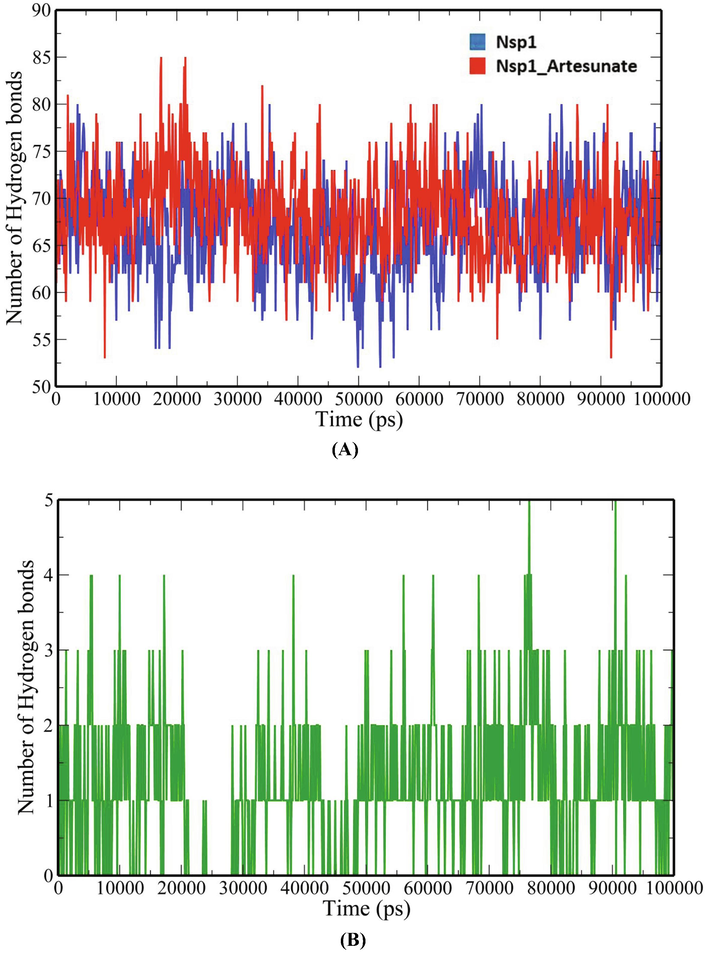
Number of hydrogen bonds-(A) intraprotein (B) Nsp1 and Artesunate.
4 Discussion
The rapid COVID-19 infection around the world warrants the need for new therapeutic antiviral agents. The discovery of a therapeutic agent is possible due to a better understanding of the disease pathogenesis. COVID-19 is caused by a betacoronavirus known as SARS-CoV-2 which relies on several non-structural proteins (Nsp1-16) for replication within the host. SARS-CoV-2 combats the host immune defence mechanisms by synthesizing Nsp1 protein, a major virulence factor. The SARS-CoV-2 Nsp1, commonly known as the host shutdown factor, inhibits the host’s gene expression and innate immune responses. It is a promising therapeutic drug target since it suppresses the antiviral immune responses of the host. Herein, we have explored artemisinin and its derivatives as possible inhibitors of SARS-CoV-2 through molecular modelling techniques. Artemisinin (ARS) is a low molecular weight bioactive compound originally isolated from the traditional Chinese medicinal plant Artemisia annua L (Xia et al., 2020). Its chemical structure, which is a sesquiterpene lactone with a peroxide bridge, has been reported to have strong antimalarial effects (Chang, 2016; White et al., 2015). It also shows additional pharmacological benefits beyond antimalarial, such as anti-virus, anti-neoplastic, anti-inflammatory, and immunosuppressive properties (An et al., 2017). Artemisinin and its derivatives (ARTs) have been recently explored for their capacity to fight SARS-CoV-2 infection due to their anti-inflammatory, immunoregulatory, and broad-spectrum antiviral characteristics (Kshirsagar and Rao, 2021). We have used a high-resolution X-ray crystal structure of the globular N-terminal domain of SARS-CoV-2 Nsp1 in this study (Semper et al., 2021). Artemisinin and its derivatives bind to the target enzyme with favourable binding energies and the interactions are mediated through hydrogen bonds and hydrophobic interactions. The three top-ranked leads identified in the studies were Artesunate, Artemiside and Artemisone which demonstrated higher binding affinities to Nsp1 than the reference drug. The compounds show favourable drug-like properties. The molecular dynamics simulations of Nsp1_Artesunate complex and unbound Nsp1 protein reveal that Artesunate significantly caused conformational changes in the target protein and stably bonded to it via hydrogen bond interactions. Nair et al. (2021) demonstrated the in vitro activity of hot water extracts of A. annua against SARS-CoV-2 and the UK (B.1.1.7) and South African variants (B.1.351) using Vero E6 and Calu-3 cells. Recently, Zhou et al. (2021) evaluated the anti-SARS-CoV-2 properties of A. annua extracts along with artemisinin, artemether and artesunate in different cell types and found artesunate to be the most potent drug candidate that targets SARS-CoV-2 at the post-entry-level. Artesunate is an antimalarial drug that is semi-synthetically prepared from artemisinin and is considered to be a promising therapy for COVID-19 because of its anti-inflammatory properties, an NF-B (Nuclear Factor kappa B)-coronavirus action, and a chloroquine-like endocytosis inhibitory mechanism (Uzun and Toptas, 2020). Cao et al. (2020) recently explored the antiviral activities of nine artemisinin-related compounds in vitro against SARS-CoV-2 and reported Artesunate, Arteannuin B, and Lumefantrine as promising anti-SARS-CoV-2 agents.
5 Conclusion
Molecular modelling approaches were used to investigate artemisinin and its derivatives as inhibitors of SARS-CoV-2 Nsp1, a key virulence factor suppressing the host's immunological responses. The study shows that Artemisinin and its derivatives including Artesunate, Artemiside, and Artemisone bind to Nsp1 with high binding affinity and establishes hydrogen bonds and hydrophobic interactions with key residues. These molecules possess favourable drug-like properties and pharmacokinetic characteristics. The binding of the best-ranked molecule, Artesunate modulates the activity of Nsp1 by lowering the flexibility of the backbone atoms, reducing the structural compactness, increasing the solvent accessible surface area and boosting the intramolecular hydrogen bond interactions. Thus, artemisinin and its derivatives hold a great promise in developing into new antiviral treatments against SARS-CoV-2.
Acknowledgements
The authors would like to extend their sincere appreciation to the Researchers Supporting Project number (RSP-2021/306), King Saud University, Riyadh, Saudi Arabia.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- COVID-19 pandemic: an overview of epidemiology, pathogenesis, diagnostics and potential vaccines and therapeutics. Ther. Deliv.. 2020;11:245-268.
- [Google Scholar]
- Antimalarial drugs as immune modulators: new mechanisms for old drugs. Annu. Rev. Med.. 2017;68:317-330.
- [Google Scholar]
- Anti-SARS-CoV-2 potential of artemisinins in vitro. ACS Infect. Dis.. 2020;6:2524-2531.
- [Google Scholar]
- The discovery of Qinghaosu (artemisinin) as an effective anti-malaria drug: a unique China story. Sci. China Life Sci.. 2016;59:81-88.
- [Google Scholar]
- SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.. 2017;7:42717.
- [CrossRef] [Google Scholar]
- DeLano, W.L., 2002. The PyMOL Molecular Graphics System. http://www.pymol.org.
- The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol.. 2020;5:536-544.
- [Google Scholar]
- Alphacoronavirus transmissible gastroenteritis virus nsp1 protein suppresses protein translation in mammalian cells and in cell-free HeLa cell extracts but not in rabbit reticulocyte lysate. J. Virol.. 2011;85:638-643.
- [Google Scholar]
- SARS coronavirus nsp1 protein induces template-dependent endonucleolytic cleavage of mRNAs: viral mRNAs are resistant to nsp1-induced RNA cleavage. PLoS Pathog.. 2011;7:e1002433.
- [Google Scholar]
- Identification of residues of SARS-CoV nsp1 that differentially affect inhibition of gene expression and antiviral signaling. PLoS One. 2013;8:e62416.
- [Google Scholar]
- A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat. Struct. Mol. Biol.. 2009;16:1134-1140.
- [Google Scholar]
- PubChem substance and compound databases. Nucleic Acids Res. 2016;44:D1202-D1213.
- [CrossRef] [Google Scholar]
- Antiviral and immunomodulation effects of Artemisia. Medicina (B. Aires). 2021;57:217.
- [CrossRef] [Google Scholar]
- LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model.. 2011;51:2778-2786.
- [CrossRef] [Google Scholar]
- Severe acute respiratory syndrome coronavirus protein nsp1 is a novel eukaryotic translation inhibitor that represses multiple steps of translation initiation. J. Virol.. 2012;86:13598-13608.
- [Google Scholar]
- SARS-CoV-2 nsp1: bioinformatics, potential structural and functional features, and implications for drug/vaccine designs. Front. Microbiol. 2020:11.
- [Google Scholar]
- Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem.. 1998;19:1639-1662.
- [Google Scholar]
- AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem.. 2009;30:2785-2791.
- [CrossRef] [Google Scholar]
- Artemisia annua L. extracts inhibit the in vitro replication of SARS-CoV-2 and two of its variants. J. Ethnopharmacol.. 2021;274:114016.
- [CrossRef] [Google Scholar]
- Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J. Virol.. 2008;82:4471-4479.
- [Google Scholar]
- Crystal structure and pair potentials: A molecular-dynamics study. Phys. Rev. Lett.. 1980;45:1196-1199.
- [Google Scholar]
- Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol.. 1993;234:779-815.
- [Google Scholar]
- PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr.. 2004;60:1355-1363.
- [Google Scholar]
- Structural characterization of nonstructural protein 1 from SARS-CoV-2. IScience. 2021;24:101903.
- [Google Scholar]
- Severe acute respiratory syndrome coronavirus nsp1 facilitates efficient propagation in cells through a specific translational shutoff of host mRNA. J. Virol.. 2012;86:11128-11137.
- [Google Scholar]
- Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science (80-). 2020;369:1249-1255.
- [Google Scholar]
- Suppression of host gene expression by nsp1 proteins of group 2 bat coronaviruses. J. Virol.. 2009;83:5282-5288.
- [Google Scholar]
- Artesunate: could be an alternative drug to chloroquine in COVID-19 treatment? Chin. Med.. 2020;15:1-4.
- [Google Scholar]
- Arbidol: A potential antiviral drug for the treatment of SARS-CoV-2 by blocking trimerization of the spike glycoprotein. Int. J. Antimicrob. Agents. 2020;56:105998.
- [Google Scholar]
- Structure of the SARS-CoV-2 Nsp1/5′-Untranslated region complex and implications for potential therapeutic targets, a vaccine, and virulence. J. Phys. Chem. Lett.. 2020;11:9659-9668.
- [Google Scholar]
- The genetic sequence, origin, and diagnosis of SARS-CoV-2. Eur. J. Clin. Microbiol. Infect. Dis.. 2020;39:1629-1635.
- [Google Scholar]
- Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J. Virol.. 2007;81:11620-11633.
- [Google Scholar]
- Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B. 2020;10:766-788.
- [Google Scholar]
- The therapeutic effect of artemisinin and its derivatives in kidney disease. Front. Pharmacol.. 2020;11:380.
- [Google Scholar]
- A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020:1-4.
- [Google Scholar]
- In vitro efficacy of Artemisinin-based treatments against SARS-CoV-2. Sci. Rep.. 2021;11:1-14.
- [Google Scholar]




