

Translate this page into:
An in-silico investigation of potential natural polyphenols for the targeting of COVID main protease inhibitor
⁎Corresponding author at: Department of Biology, College of Science, Princess Nourah bint Abdulrahman University, Riyadh 13415, Saudi Arabia. nhaljarba@pnu.edu.sa (Nada H. Aljarba)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The deadliest recent pandemic outbreak of COVID-19 disease has severely damaged the socio-economic health of the people globally. Due to unavailability of any effective vaccine or treatment the human beings are still struggling to overcome the pandemic condition. In an attempt to discover anti-COVID molecule, we used in-silico approach and reported 160 natural polyphenols to identify the most promising druggable HITs that can further used for drug discovery process. The co-crystallized structure COVID protease enzyme (PDB id 6LU7) was used. HTVS, MD simulation, binding energy calculations and in-silico ADME calculation were done and analyzed. Depending upon the scores three compounds galangin, nalsudaldain and rhamnezine were identified and the docking score were found to be −7.704, −6.51, −4.212 respectively. These docked complexes were further subjected to MD simulation runs over a 100 ns time and the RMSD and RMSF values were determined. The RMSD values of three compounds were found to be 2.9 Å, 7.6 Å & 9.5 Å respectively and the lowest RMSF values suggested the steady stability of ligand–protein complexes. The binding free energies (ΔG) of compounds with protein were found to be −49.8, −56.45, −62.87 kJ/mole. Moreover, in-silico ADME calculations indicated the drug likeliness properties of these molecules. By considering all these in-silico results the identified HITs would be the most probable anti-COVID drug molecules that can be further taken in wet lab and can act as lead for development of newer inhibitor of COVID-19 main protease enzyme.
Keywords
COVID-19
Mpro
Polyphenols
HTVS
MD simulations

1 Introduction
The newly reported COVID-19 disease in China province Wuhan has been affected billions of people and causing millions of deaths worldwide since its outbreak. Due to its occurrence and nature, WHO declared it as pandemic in March 2020 (Cucinotta and Vanelli, 2020). Till date the number of cases reported and death associated with COVID-19 is continuously increasing and very badly affecting the socio-economic health of developed as well as developing countries (Mahase, 2020). The different variants and mutations in COVID-19 virus have stuck the world and global health system to think and rethink about how to tackle or control the current pandemic situation. The common symptoms are varied in different cases reported by different variants or mutated virus that also worrying the scientist and physician to diagnose and treat the patient. The observed common symptoms in COVID patients may be absent or present in different mutated viruses (Li et al., 2020; Pan et al., 2020). The epidemiological studies revealed that the COVID-19 virus spreadability of the virus is higher (2–2.5%) and fatality is lower (5%) compare to other earlier reported corona viruses SARS, MERS that had the spreadability and fatality rate of 1.7–1.9% & 9.5% and <1% & 34.4% respectively (WHO, 2019; Ahmadzadeh et al., 2020; Deng et al., 2020; To et al., 2020). The RT-qPCR test from COVID-19 patients showed the highest viral load during the first week of onset of infection (Chakraborty et al., 2020; Tang et al., 2020; Zhai et al., 2020). Then after 20 days the median viral load was observed and antibody production was started from the 10th day of viral infection. Cohort study of COVID-19 patients indicated the old age and patients with comorbidities are more prone or susceptible to severe action of infection and high risk of death. The detailed epidemiology, diagnostic and treatment options have been reported. FDA has given the emergency approval of few monoclonal antibodies and antiviral molecule for the uncontrolled spread of COVID-19 infection. Newer antiviral molecules such as Nirmatrelvir with ritonavir, Remdesivir and Molnupravir. From investigational monoclonal antibodies Bebtelovimab was also considered for emergency use in adults and children above 12 years. (Chakraborty et al., 2020; Ejaz et al., 2020; Bajgain et al., 2021; Mithal et al., 2021). After the outbreak various existed drug candidate have been tried as drug repurposing for possible use in COVID-19 infection. As per the Coronavirus Treatment Accelerated Program (CTAP) developed and maintained by Food and Drug Administration (FDA) more than 570 drug candidate are being monitored for drug development process (Mandour et al., 2020; Touret et al., 2020). The recent reports suggested the drugs targeting viral replication mechanism are of major attention for possible use in COVID-19 (Biering et al., 2021). The ritonavir–lopinavir drug combination have shown the significant improvement when administered in hospitalized patient in China. However, the efficacy of these drugs remains questionable and insignificant (Javorac et al., 2020; Marcolino et al., 2020; Stasi et al., 2020; Trezza et al., 2020; Verdugo-Paiva et al., 2020).
Proteases are the enzymes that play the important role in viral life cycles. They are responsible for the cleaving of polyprotein and continuing the viral replication. The infectious viruses encode one more protease that has the important role in viral life cycle. Thus, these proteases (3CLpro or Mpro) provide the genetically assured therapeutic target for successful treatment of viral infection (Tong, 2002; Roe et al., 2021). In resistance or mutated cases, the protease inhibitors were given in combination with other antiviral drugs. The functional polypeptide needed for transcription and replication i.e. pp1a and pp1ab were released by conserved catalytic activity of 3CLpro. These polypeptides are encoded by the SARS-COV-2 enzyme. This important cleavage activity of 3CLpro in virus replication process as well as absence of its homolog in human being led to the important target for development of anti-COVID drug candidate. Various researcher has been explored this target but the question for its important utility as an antiviral drug development still remains unsuccessful and it need to be explored (Hilgenfeld, 2014; Wang et al., 2020, Amin et al., 2021). Even many promising drug candidates failed in advance stages of drug discovery processes due to less credibility of preliminary data. Hence rational approach like Computer Aided Drug Design (CADD) have been proven to be a powerful tool and used since last few decades (Leelananda and Lindert, 2016; Romano and Tatonetti, 2019; Sonawane et al., 2019). High Throughput Virtual screening (HTVS), drug-ligand interaction, MD simulation, binding free energy calculations, in-silico ADME calculations are the computational approach used to identify the drug candidate from the set of existing chemical data base. The credibility of in-silico data generated via above method are the helpful for judging and proceeding the identified HIT into further stages of drug discovery processes. Many researchers have been used these computational tools for the successful identification of small molecule inhibitors against different receptors (Bagchi et al., 2017; Usman et al., 2018; Siddique et al., 2020; Wang, 2020). The co-crystal structure of SARS CoV protease (Clpro) with N3 inhibitor was reported by Jin et al (PDB id: 6LU7). In this study the computational approach docking and MD simulation have been done and binding energy with stability of complex was analyzed.
2 Material and methods
The proposed work include the computational approach for identification of newer COVID-19 main protease inhibitor from the natural polyphenols. The adopted methodology was depicted in Fig. 1. A total 160 natural phenolic compounds were initially docked at the substrate binding pocket of COVID-19 main protease enzyme using the available co-crystallized structure (PDB id 6LU7). The obtained top 3 Hits were further subjected to MD simulations and free binding calculation followed by in-silico prediction of ADME properties.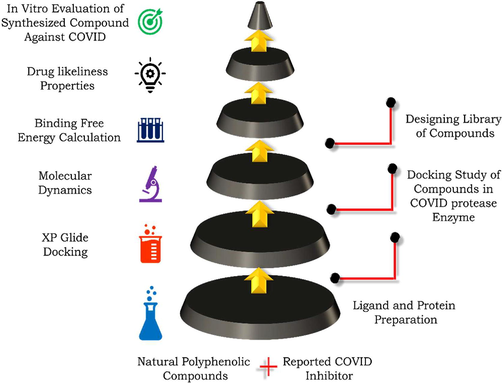
Workflow diagram from bottom to top for the identification of COVID-19 main protease enzyme from natural polyphenols.
2.1 Protein preparation
The co-crystal structure of COVID-19 with inhibitor N3 was downloaded from the public repository protein data bank (Liu et al., 2020). The protein was prepared by using protein preparation wizard. The water molecules beyond the 5 Å were deleted and H atoms were added. If heteroatom is present in the protein chain, then using Epik module the heterostate was generated at pH 7 ± 0.3. The amide groups were re-oriented to aspartic and glutamic acid. The H-bonds were optimized using OPLS2005 and energy minimization was done to avoid any interactions. Then by using receptor grid generation wizard the grid was prepared using the default parameters by selecting the internal ligand to specify the binding site. The conformers, tautomer and rotamers were generated for the molecules. Energy minimization was done and these prepared ligands were used for docking studies. For docking the software was validated by extracting and redocking the internal ligand at the active site and the RMSD value was calculated. The centroid of the co-crystalline ligand was specified for docking of the selected compounds. The post docking minimization was performed and 5 poses per ligand interaction was recorded. Maestro pose viewer and Glide XP visualizer was analyzed the best docking pose (Schrodinger, 2011).
2.2 Molecular dynamics simulation
The best docking and protein docked complex were used for molecular dynamics simulation studies. Desmond module developed by DE Shaw group was used for molecular dynamics simulation studies. System builder panel was used to generate the water solvated orthorhombic simulation box. The box was prepared with the Simple Point-Charge (SPC) explicit water model and minimum 10 Å distance was maintained between the solvent and protein surface. The counter ion was used to neutralized the solvated system with 0.15 M concentration of physiological salts. The prepared system was put for energy minimization by using Macromodel tool. Following this, the RESPA integrator, Martyna-Tobias-Keilnbarostat and Nose-Hoover chain thermostat algorithm with 2 ps was used as relaxation time for ligand–protein complex before simulation. This system was considered for MD simulation studies for 100 ns time. It was run at 310 K & pressure 1.0 bar with isothermal-isobaric ensembles means constant temperature, pressure and particles with default setting in Desmond module. The prepared and energy minimized system then was used for MD run over 100 ns trajectory or for the time specified in individual study. After the successful run for viewing and the movie was generated by using the -out.cms file. Moreover, the trajectory during MD run was written with 100 frames and understand the protein–ligand complex stability during MD run, the initial and protein backbone frames were aligned. Finally, the RMSD and RMSF plots were obtained by -outacts file through simulation interaction diagram (Bergdorf et al., 2015).
2.3 MM-GBSA screening
MM-GBSA is used to re-score the results obtained by docking HTVS. The 10% dataset was used for to recheck the credibility of such results. In this method the top scoring pose with top 10 best docked pose of every molecule was used and binding free for each drug-ligand complex were calculated. Here, we used the molecular dynamics based lowest energy complex for the calculation of minimum energy. This procedure involved the MD simulation of 100 ps and the lowest energy minimized complex was used for MM/GBSA calculations (Mali and Chaudhari, 2018).
2.4 In-silico ADME calculations
To avoid the later stage failure of drug discovery process and to check the drug likeliness properties of molecules the in-silico ADME calculation was done. The pharmacokinetic profile was checked and the druggability of the molecule was assessed. In present study we used the QikProp v3.0 a Schrödinger utility to calculate the ADME parameters. These calculations were generally based on the Lipinski’s rule of five (Ro5) and Jorgensen Rule of 3. The various physicochemical parameters involved in this study were generally responsible for good pharmacokinetic behavior. The common physicochemical properties considered are molecular weight, logP, H bond donor and acceptor, lipophilicity, hydrophilicity, human oral absorption and Caco-2 cell permeability etc.
3 Results
3.1 3CL protease as antiviral drug target
Protease is considered to be crucial target for design of antiviral drugs. The co-crystal structure of 3CLpro SARS COV-19 enzyme with inhibitor N3 was reported. The structure reported the presence of single polypeptide as unsymmetrical unit. The two protomer (A & B) of these polypeptides known to form an axis symmetry. Each protomer consist of three domains and each of these domains I to III contain residues ranging from 8 to 101, 102–184 and 201–303 respectively. The domain-III comprises of five α-helices which are assembled in antiparallel globular cluster. The domain II and III are interconnected by along loop region. Cys-His catalytic dyad is present in SARS-CoV-2 Mpro and in between Domain-I and II the substrate binding site is present which are similar to previously reported other types of coronaviruses. The compound N3 was present in the cocrystal structure (PDB id: 6LU7) used to study the molecular modelling study. The 50% cytotoxicity of compound N3 was <133 μM and compound N3 was found to be specific inhibitor of Mpro enzyme of various coronaviruses including MERS and SARS. The docking result showed the molecule actively binds at the substrate binding site and was found to be time dependent irreversible inhibitor of SARS-COV Mpro enzyme in kinetic analysis. For the better understanding of interactions between N3 and SARS-CoV, the various active sites present in N3 were named as P1, P2, P3, P4 & P5 (Fig. 2). The compound N3 displayed the multiple H bonds with various residues of S2 subsites that is responsible for the locking of inhibitor into the substrate binding pocket. The amino acid residues H1, M49, Y54 & M165 present at subsite S2 of active pocket formed the hydrophobic interactions with N3. While site P3 present in N3 was exposed to solvent means we can optimize this site with various functional groups to alter the physicochemical properties with the extent of our need. The P4 site interacted with F85, Q192, L167 and M165 active site residues present at protomer A. The P168 residue of protomer A and 190–191 backbone residues displayed the interaction with site P5 of N3. This study also revealed that the domain-III, loop surface and substrate binding pocket are highly conserved in 3CL Mpro co-crystal structure. Thus, extent of active fitting and binding of molecules with these regions could be prerequisite for specificity and potency of inhibitor as an anti-COVID molecule. The same study also reported the ebselen as a most promising anti-COVID molecule with IC50 value of 1.55 ± 0.3 μM (Jin et al., 2020).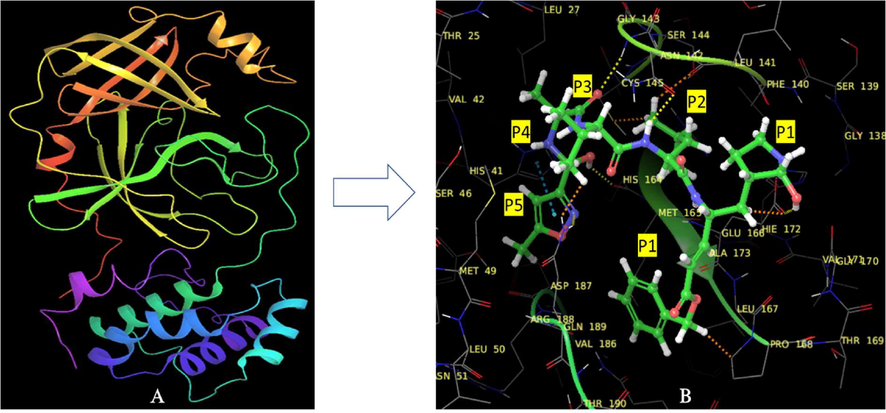
3D crystallographic structure of COVID main protease enzyme (pdb id: 6LU7) b) the internal co-crystallized ligand with its different sites (P1-P5) as reported by Jin et al.
3.2 Molecular docking
The present work used the ebselen as a reference. The obtained results were analyzed and compared with the reported N3 molecule to generate the most creditable in-silico data. Before start of docking the software was validated by redocking of internal ligand and obtained RMSD value 1.6841 Å indicated the software could be used for further study. Figs. 3 and 4 displayed the 3D and 2D interactive diagram of reference molecule (A), alangin (B), nalsudaldain (C) and rhamnezine (D), respectively. The various interactions of these four molecules were summarized in Table 1. The reference molecule displayed the polar H bond with Gly143 of protomer and hydrophobic interactions with Met165, Met49, Leu27 & Cys145. The π- π stacking interaction was displayed by His143. The polar interactions with Thr25, Thr26, His41, His146, Gln189, Asn142 and Ser144. As with compound N3 the H bond helped in locking of molecule whereas other interactions are responsible for the stabilization of complex. Followed by this the other phenolic compound galangin showed the promising docking score with −4.34. The similar with ebeselen reference molecule galangin also formed the H-bond with His41 of domain I. Whereas nalsudaldain formed the 3H-bonds with Gly143, Glu166, Gln189. This molecule had two more additional H-bond than reference molecule. Unfortunately, the fourth molecule rhamnezine formed the H bond with other residues Thr46. The molecule did not leave the active pocket but occupied the same pocket as with esbselen and displayed the hydrophobic interactions similar. The P4 site of N3 molecule displayed the hydrophobic interaction with Met165, Leu167, F85 and Q192 present in protomer A. Fortunately, all the 3 phenolic compounds and ebselen molecule retained the same active pocket by displaying the hydrophobic interactions with common residues. If we could see, galangin molecule Leu27, Val42, Met49 residues of domain-I and Leu141, Cys145 & Met165 from domain -II displayed the hydrophobic interaction. The other molecule nalsudaldain actively bind with the substrate binding pocket with the docking score of −6.026. It displayed the hydrophobic interactions with Leu27, Val42, Met49, Leu141, Met165, Leu167 and Pro168 residues. The polar interactions with His41, Ser46, Glu189, His164, Asn142, Thr26, Thr25 active site residues. The compound galangin and nalsudaldain displayed the polar interactions with backbone residue Thr190 of protomer A and this interaction was also found with the reported N3 compound. However, both the molecules had polar interactions with other amino acid residues His41, Glu189, Thr190, Thr26, Thr25, Asn142, His164 from the same subsite. Thus, we analyzed the various interactions of all the top three phenolic compound and reference molecule ebselen and this further compared with co-crystallized ligand N3 to confirm the obtained results. And we consider that the identified top three phenolic compounds bound and fitted with the substrate biding pocket in a similar fashion with N3 and ebselen molecule. The in-vitro antiviral activity of both the compound had been proven as potent molecule. We thought these three molecules would be the potential candidate against SARS corona viruses as they interacted with similar active site residues with that of N3 and ebselen. Hence, we took these molecules further confirmation and generation of more validated in-silico data.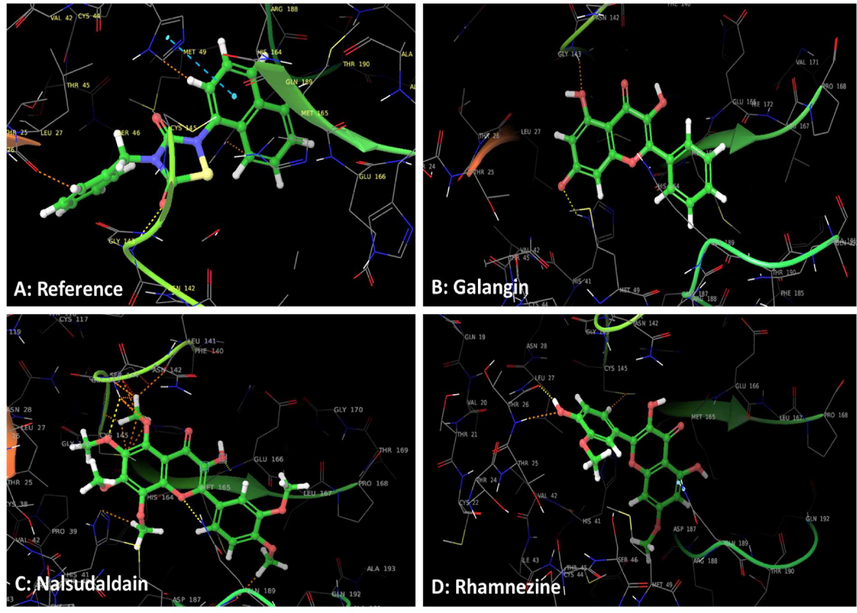
Overlay of 3D interactive diagram of reference molecule (A), galangin (B), nalsudaldain (C) and rhamnezine (D). The molecules were represented by green ball and stick model, dotted yellow line indicate the H bond and brown dotted line show the H bond with backbone residues, whereas sky blue line represent the p-p staking interactions.
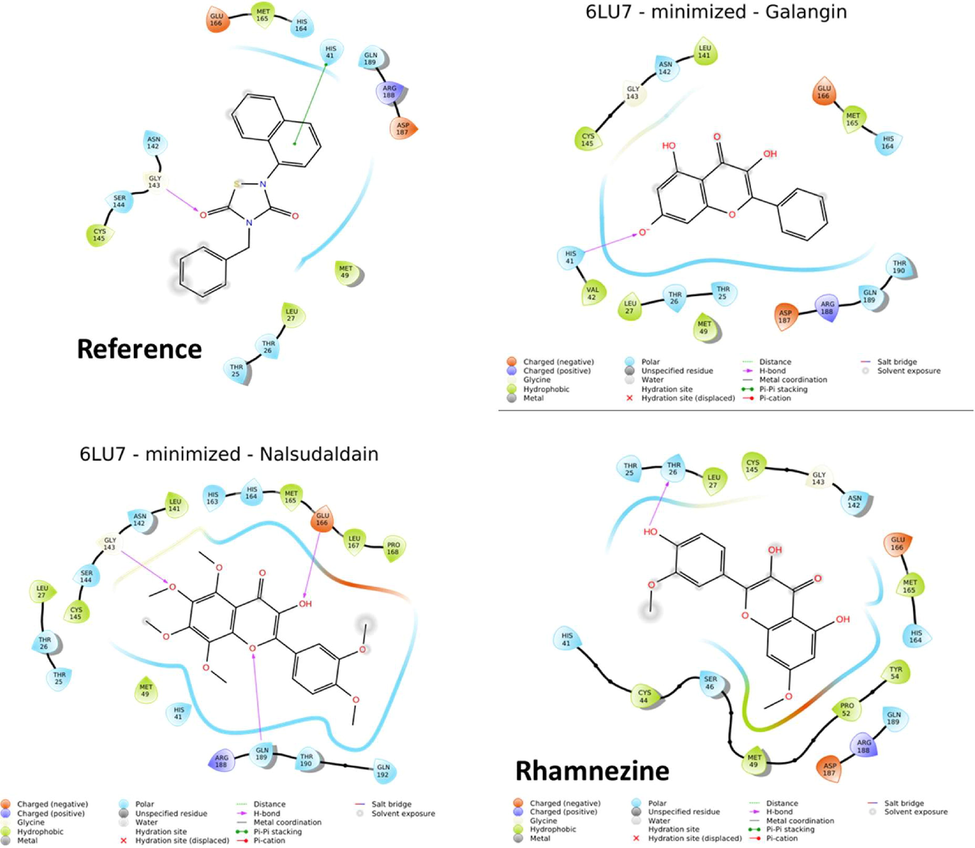
2D interactive diagram of reference molecule. Different colors of amino acid residues indicate the various types of interactions that is written at the bottom of figure.
Sr No.
Molecule
Docking score
Type of interactions
H bond
Hydrophobic
Polar
1.
Reference
(Ebselen)−4.21
Gly143
Met165, Met49, Leu27 & Cys145
Thr25, Thr26, His41, His146, Gln189, Asn142 and Ser144
2.
Rhamnezine
−6.23
–
Leu27, Cys145, Cys44, Met49, Met165, Thr54, Pro52 and Met
His41, Ser46, Glu189, His164, Asn142, Thr26, Thr25
3.
Nalsuldaldain
−6.02
Gly143, Glu166, Gln189
Leu27, Val42, Met49, Leu141, Met165, Leu167 and Pro168
His41, Glu189, Thr190, Gln192, Thr26, Thr25, Ser144, Asn142, His163, His164
4.
Galangin
−4.34
His41
Leu27, Val42, Met49, Leu141, Cys145 & Met165
His41, Glu189, Thr190, Thr26, Thr25, Asn142, His164
3.3 MD simulations studies
To validate the molecular docking results the active ligand–protein complexes were further subjected to molecular dynamics studies. All three identified phenolic were individually subjected to MD simulation run for 100 ns trajectory. The comparative stability of all three ligand–protein complexes was analyzed by their derivation and fluctuations. The various % of various interactions over the entire 100 ns were also considered for results analysis. The binding free energies (ΔG) of docked drug-ligand complexes were also calculated and the molecules were ranked according to lowest their lowest binding energy scores.
3.4 RMSD and RMSF
The root mean square derivation and fluctuations were calculated by the backbone RMSD and fluctuation performed using the ligand–protein complex over 100 ns run and it was depicted in Figs. 5 and 6 respectively. The calculated RMSD indicated the simulated galangin complex attained the steady stability after the 9 ns time of run. Again, at the 70 ns of run time the simulated complex deviate slightly but again it maintained the stability after 80 ns. The nalsudaldain simulated complex maintained the steady stability within a 5 ns of run time and it carried for 95 ns. At the last time of run the complex displayed the slight deviation. Whereas, rhamnezine complex quickly maintained the stability within 4 ns run time and maintained over 100 ns MD simulation run. The average RMSD values for galangin, nalsudaldain and rhamnezine simulated complexes were found to be 7.6 Å, 2.9 Å and 9.5 Å respectively. RMSF parameter was used to determined flexible and rigid site of the protein. Further various interactions from docked ligand complexes and 100 ns simulated complexes were compared and analyzed and it was depicted in Fig. 7. The galanginin docked pose displayed the one H-bond with His41. Apart from this there was three new H-bonds were observed with Thr190, Arg188 and Gln 192 for a period of 69%, 58% and 50% respectively as shown in the Fig. 7. The same complex maintained the hydrophobic interactions with Asp187, Gln189 and Gln192. Similarly, the second molecule nalsudaldain can retained the H bond interactions with Gly143 for a period of 78% run time of 100 ns MD trajectory. However, the simulated complex displayed the two more H-bond interactions His41 and Ser144 for a period of 71% and 42% respectively. The third complex rhamnezine displayed the H-bond interaction with Thr26 for a very short period but with other three residues Thr190, Gln192 and Arg188 the rhamnezine molecule displayed a very good H-bond interaction for 89%, 72% and 58% respectively over 100 ns run. Moreover, the hydrophobic interactions were also retained in rhamnezine-protein complex as shown in Fig. 7.
Display of RMSD value of identified compounds over 100 ns trajectory.
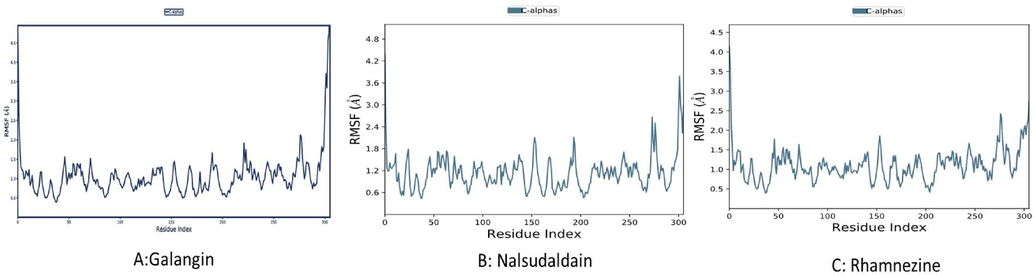
Display of Root mean square fluctuations of the residues over entire 100 ns trajectory.
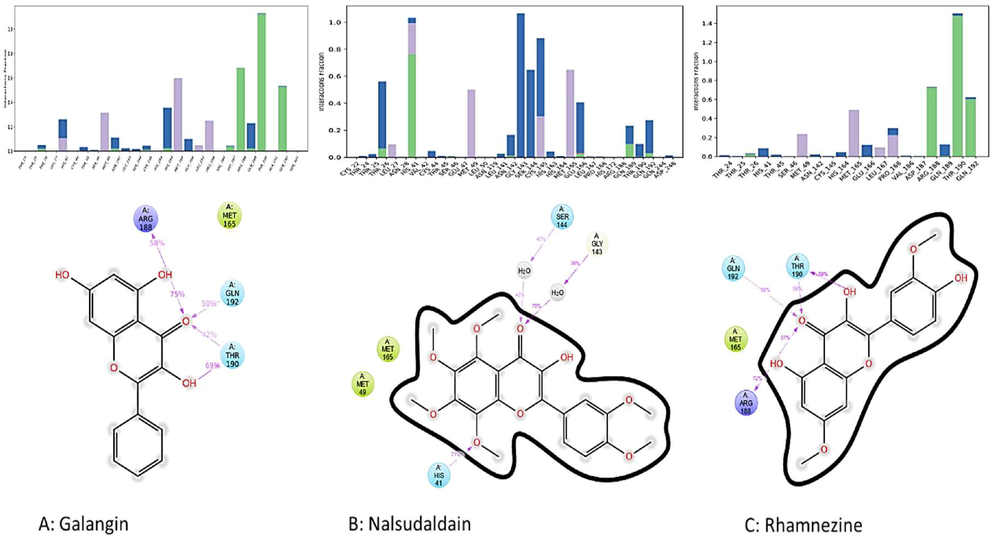
Different interaction conserved or displayed by the molecule after the 100 ns run MD simulation.
3.5 Calculation of binding free energies (ΔG)
The binding affinities can be calculated by the molecular docking studies but it lacks the suitable use of flexibility hence many times it gave inconsistent data. Hence, more trusted molecular mechanics tool MM/GBSA (Poisson–Boltzmann surface area) was used to calculate binding free energies of ligand–protein complex. All four active ligand–protein complexes were subjected to Prime MM/GBSA calculation and the results were summarized in Table 2. The nalsudaldain-protein complex was found to have a lowest binding energy and said to be most stable complex. We can say the binding affinities will be more in this complex. However, galangin-drug complex was shown least binding energy. Comparatively amongst all four drug-protein complexes, this complex is least stable or have least binding affinity (see Fig. 8).
Sr No.
Molecule
Prime Energy
(ΔG) (kJ/mole)Prime dG bind
ΔG (kJ/mole)
1.
Ebselen
−13414.22
−56.45
2.
Galangin
−13386.98
−49.8
3.
Nalsudaldain
−13423.68
−69.41
4.
Rhamnezine
−13406.01
−62.87
Proposed outcome of the study.
3.6 In-silico ADME calculation
Physicochemical properties of any drug molecule play crucial role with respect to its pharmacokinetic profiling. This should be taken into consideration in early stages of drug design process. As later stage failure of promising druggable candidate due to poor pharmacokinetic behavior will cost a huge loss in terms of money and time. In-silico prediction of ADME properties of identified HIT before it proceeds in an advance stage of would avoid the failure of drug discovery process. Hence, here also the identified phenolic compounds with reference molecule ebselen was subjected to in-silico ADME calculation using qikprop utility. The results obtained were written in Table 3. The various parameters such as molecular weight, Lipinskie’s rule of 5 i.e Ro5, Ro3, partition coefficient, metabolism, HB donor, HB acceptor and logBB (Blood brain barrier). The obtained values indicated the all identified HITs have the significant ADME properties and it will absorb orally with good tolerance rate. The values also suggested about the metabolic fate of identified HITs. The molecules were not violated the Lipinskie’s rule of 5 and rule of 3 indicating the druggable properties of the molecules. The LogBB value indicated the all molecules would not cross the blood brain barrier and cannot produce CNS toxicities. With the help of these values, we determined the pharmacokinetic behavior of all three identified HITs such as galangin, nalsudaldain and rhamnezine. By using these in-silico values, we can postulate that the molecules can absorb orally, distributed well in the body and reach to specific site in unaltered form to produce the desired effect.
Sr No.
Parameters
Ebselen
Nalsudaldain
Rhamnezine
Galangin
1.
HB Donor
0
1
2
2
2.
HB Acceptor
3.5
7.75
5.25
3.75
3.
LogPO/W
4.285
3.193
2.146
1.769
4.
logBB
−0.150
−0.494
−1.284
−1.195
5.
% Human oral Absorption
100
100
83.204
78.385
6.
PSA
56.725
94.639
108.768
94.832
7.
Ro5 Violation
0
0
0
0
8.
Ro3 Violation
0
1
0
0
9.
Molecular Weight
334.39
418.39
330.29
270.24
4 Discussion
The wise and intelligent decision is needed to choose the suitable candidate which is taken into advance stages of drug design project. To select any molecule many organizations are using computational tool to simulate the wet lab conditions and generating the in-silico data. Based on these in-silico data the fate of molecules is decided whether it is to be taken into later stage or not. But after generating so many in-silico data, still many promising druggable candidate failed in later stages and here the questions on credibility of in-silico data come. Hence, combinations of different computational tools are supposed to be useful to generate the more credible in-silico data which can be considered to decide the suitable drug molecule for advance stages of drug design process (Makhouri and Ghasemi, 2018, Brogi et al., 2020). Different groups used the different set of computational tools towards the rational approach-based drug design including HTVS, molecular modelling, dynamic simulations studies, ADME, metabolic studies, pharmacophore mapping and QSAR studies (Bagchi et al., 2017; Siddique et al., 2018a; Siddique et al., 2020; Mary et al., 2021). The right combinations and validated in-silico data could help to choose the potential candidate for the advance drug discovery process. In this study we used the HTVS, molecular modelling, dynamics simulations and binding free energy calculations of natural polyphenolic. By considering the fact that these tools would generate the more validated in-silico data against the COVID-19 main protease enzyme and identified HITs can be used for development newer molecule which can be used for newer pandemic. The HTVS study was performed using the naturally occurring polyphenolic compounds and COVID main protease. The results thus obtained is summarized in Table 1. As with compound N3 the H bond helped in locking of molecule whereas other interactions are responsible for the stabilization of complex (Liu et al., 2020). The galangin also formed the H-bond with His41 of domain I. whereas nalsudaldain formed the 3H-bonds with Gly143, Glu166, Gln189. This molecule had two more additional H-bond than reference molecule. The P4 site of N3 molecule displayed the hydrophobic interaction with Met165, Leu167, F85 and Q192 present in protomer A. Fortunately, all the 3 phenolic compounds and ebselen molecule retained the same active pocket by displaying the hydrophobic interactions with common residues. The in-vitro antiviral activity of both the compound had been proven as potent molecule. We thought these three molecules would be the potential candidate against SARS corona viruses as they interacted with similar active site residues with that of N3 and ebselen. Hence these molecules were considered for further dynamic simulation studies to see the stability of ligand–protein complex. RMSD and RSMF values were used to check the stability of docked ligand protein complex and lower RMSD value indicate the stability and RMSF value suggested the molecule did not fluctuate from the active pocket of COVID main protease active pocket site. The obtained results were displayed in Figs. 5 & 6. Also, we determined the RMSF values of all three phenolic compounds. It was observed that none of the molecule significantly fluctuated over the entire run of MD trajectory. These RMSD and RMSF values suggested that all three simulated complexes were quite stable and there were no deviation or fluctuations observed. To validate the molecular modelling studies binding free energy calculations of identified HITs and reference molecules were performed. As the study reported about link between binding free energy could be used to validate the molecular docking studies and here also, we done the same thing. The docked drug-ligand complexes were subjected to binding free energies calculations and the results obtained were written in Table 2. Compared to reference molecule galangin drug complex was shown least binding energy. Moreover, the pharmacokinetic properties of drug molecule are always of major concern and many drug molecules failed because of poor pharmacokinetic profiling. The drug likeliness properties of identified HITs and reference molecule were determined by in-silico ADME calculations and the results were tabulated in Table 3. None of the molecule violate the Ro3 and Ro5 for drug likeliness except nalsudaldain. But other properties remained similar and comparative and falls under the drug likeliness properties. The scores suggested that these molecules can absorb orally, well distributed in body and had the all druggable like properties (Darvas et al., 2002). Hence, could be taken into further stages of drug design and discovery processes.
5 Conclusion
This study considers the natural polyphenolic compounds for their probable use in corona virus infection. The rational approach of drug design and discovery processes or in order to avoid the risk of phase-II and Phase-III stage failure of drug development process, we used the HTVS approach, MD simulations studies, binding free energy calculations and in-silico ADME calculations for identification of suitable HIT against the SARS COVID-19. Taking the various advantages of natural compounds, good tolerability, lesser side effects and availability we used the various natural polyphenolic compounds to run the HTVS. The molecule with docking score were further used for in-silico calculation. The docked complexes all three ligands were subjected to MD simulation run. The simulated complexes were used for binding energy calculations. The calculated in-silico properties also suggested about the drug likeliness properties of the identified HITs. Hence the results obtained were suggested that all the three identified HITs would be the potential candidate against recent SARS corona virus infections.
6 Availability of data and materials
The data generated in this article are online publicly available without request.
7 Authors’ contributions
Nada H. Aljarba was performed the electrostatic study; Saad Alkahtani and Md Saquib Hasnain was performed the molecular docking studies. Mashael Bin-Meferij acquired and analyzed the data. Nada H. Aljarba and Mashael Bin-Meferij were performed dynamics simulations and SiteMap analysis.
Acknowledgement
The authors extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project number (PNU-DRI-Targeted-20-031).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- The risk factors associated with MERS-CoV patient fatality: a global survey. Diagnostic microbiology and infectious disease. 2020;96(3):114876
- [Google Scholar]
- Protease targeted COVID-19 drug discovery and its challenges: Insight into viral main protease (Mpro) and papain-like protease (PLpro) inhibitors. Bioorg. Med. Chem.. 2021;29:115860
- [Google Scholar]
- High throughput virtual screening based discovery of dengue protease inhibitor. Journal of Pharmaceutical Chemistry. 2017;4(3):35-40.
- [Google Scholar]
- High throughput virtual screening based discovery of dengue protease inhibitor.. 2017;4(3):35-40.
- Prevalence of comorbidities among individuals with COVID-19: A rapid review of current literature. American journal of infection. 2021;control49(2):238-246.
- [Google Scholar]
- Bergdorf, M., S. Baxter, C. A. Rendleman and D. E. Shaw (2015). “Desmond/GPU Performance as of October 2015.” DE Shaw research.
- “Screening a library of FDA-approved and bioactive compounds for antiviral activity against SARS-CoV-2.” ACS infectious. 2021;diseases7(8):2337-2351.
- In silico methods for drug design and discovery. 2020;8::612.
- SARS-CoV-2 causing pneumonia-associated respiratory disorder (COVID-19): diagnostic and proposed therapeutic options. Eur Rev Med Pharmacol Sci. 2020;24(7):4016-4026.
- [Google Scholar]
- Extensive partnership, collaboration, and teamwork is required to stop the COVID-19 outbreak. Archives of medical. 2020;research51(7):728-730.
- [Google Scholar]
- WHO declares COVID-19 a pandemic. Acta Bio Medica: Atenei Parmensis. 2020;91(1):157.
- [Google Scholar]
- Darvas, F., G. Keseru, A. Papp, G. Dorman, L. Urge and P. J. C. t. i. m. c. Krajcsi (2002). “In silico and ex silico ADME approaches for drug discovery.” 2(12): 1287-1304.
- Deng, Y., W. Liu, K. Liu, Y.-Y. Fang, J. Shang, L. Zhou, K. Wang, F. Leng, S. Wei and L. Chen (2020). “Clinical characteristics of fatal and recovered cases of coronavirus disease 2019 in Wuhan, China: a retrospective study.” Chinese medical journal133(11): 1261.
- COVID-19 and comorbidities: Deleterious impact on infected patients. Journal of Infection and Public Health. 2020;13(12):1833-1839.
- [Google Scholar]
- From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. The FEBS. 2014;journal281(18):4085-4096.
- [Google Scholar]
- An overview of safety assessment of the medicines currently used in the treatment of COVID-19 disease. Food Chem. Toxicol.. 2020;111639
- [Google Scholar]
- Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582(7811):289-293.
- [Google Scholar]
- Computational methods in drug discovery. Beilstein journal of organic. 2016;chemistry12(1):2694-2718.
- [Google Scholar]
- Li, L. q., T. Huang, Y. q. Wang, Z. p. Wang, Y. Liang, T. b. Huang, H. y. Zhang, W. Sun and Y. Wang (2020). “COVID‐19 patients' clinical characteristics, discharge rate, and fatality rate of meta‐analysis.” Journal of medical virology 92(6): 577-583.
- “The crystal structure of COVID-19 main protease in complex with an inhibitor. 2020;N3.”:10.
- Coronavirus: covid-19 has killed more people than SARS and MERS combined, despite lower case fatality rate, British Medical Journal Publishing. Group. 2020
- [Google Scholar]
- Makhouri, F. R. and J. B. J. C. T. i. M. C. Ghasemi (2018). “Combating diseases with computational strategies used for drug design and discovery.” 18(32): 2743-2773.
- Computational studies on imidazo [1, 2-a] pyridine-3-carboxamide analogues as antimycobacterial agents: Common pharmacophore generation, atom-based 3D-QSAR, molecular dynamics simulation, QikProp, molecular docking and prime MMGBSA approaches. Open Pharmaceutical Sciences Journal. 2018;5(1):12-23.
- [Google Scholar]
- A multi-stage virtual screening of FDA-approved drugs reveals potential inhibitors of SARS-CoV-2 main protease. J. Biomol. Struct. Dyn. 2020:1-12.
- [Google Scholar]
- What to expect from different drugs used in the treatment of COVID-19: A study on applications and in vivo and in vitro results. Eur. J. Pharmacol.. 2020;887:173467
- [Google Scholar]
- Mary, S. J., M. U. M. Siddique, S. Pradhan, V. Jayaprakash, C. J. S. A. P. A. M. James and B. Spectroscopy (2021). “Quantum chemical insight into molecular structure, NBO analysis of the hydrogen-bonded interactions, spectroscopic (FT–IR, FT–Raman), drug likeness and molecular docking of the novel anti COVID-19 molecule 2-[(4, 6-diaminopyrimidin-2-yl) sulfanyl]-N-(4-fluorophenyl) acetamide-dimer.” 244: 118825.
- High prevalence of diabetes and other comorbidities in hospitalized patients with COVID-19 in Delhi, India, and their association with outcomes. Diabetes & Metabolic Syndrome: Clinical Research & Reviews. 2021;15(1):169-175.
- [Google Scholar]
- Viral load of SARS-CoV-2 in clinical samples. The Lancet infectious. 2020;diseases20(4):411-412.
- [Google Scholar]
- Targeting novel structural and functional features of coronavirus protease nsp5 (3CLpro, Mpro) in the age of COVID-19. J. Gen. Virol.. 2021;001558
- [Google Scholar]
- Informatics and computational methods in natural product drug discovery: A review and perspectives. Front. Genet.. 2019;10:368.
- [Google Scholar]
- Schrodinger, L., 2011. Schrodinger software suite. New York: Schrödinger, LLC670.
- Comparative Shape and Electrostatic Study of Highly Potent and Selective CYP1B1 Inhibitor: Assessment of Active Site of CYP1B1 by Binding Mode Analysis Using Site. Map Tool. 2018;52(1):159-165.
- [Google Scholar]
- Comparative Computational Studies on Selective CytochromeP450 1B1 Inhibitors. Int. J. Bioautomat.. 2020;24(3):213-224.
- [Google Scholar]
- Comparative Computational Studies on Selective CytochromeP450 1B1 Inhibitors. 2020;24(3):213.
- Sonawane, V., M. U. M. Siddique, S. S. Jadav, B. N. Sinha, V. Jayaprakash and B. Chaudhuri (2019). “Cink4T, a quinazolinone-based dual inhibitor of Cdk4 and tubulin polymerization, identified via ligand-based virtual screening, for efficient anticancer therapy.” European journal of medicinal chemistry165: 115-132.
- Laboratory diagnosis of COVID-19: current issues and challenges. J. Clin. Microbiol.. 2020;58(6) e00512-00520
- [Google Scholar]
- Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: an observational cohort study. Lancet. Infect. Dis. 2020;20(5):565-574.
- [Google Scholar]
- In vitro screening of a FDA approved chemical library reveals potential inhibitors of SARS-CoV-2 replication. Scientific reports. 2020;10(1):1-8.
- [Google Scholar]
- An integrated drug repurposing strategy for the rapid identification of potential SARS-CoV-2 viral inhibitors. Scientific Reports. 2020;10(1):1-8.
- [Google Scholar]
- Combined protein and ligand based physicochemical aspects of molecular recognition for the discovery of CDK9 inhibitor. Gene Reports. 2018;13:212-219.
- [Google Scholar]
- Lopinavir/ritonavir for COVID-19: A living systematic review. Medwave. 2020;20(06):e7966.
- [Google Scholar]
- Fast identification of possible drug treatment of coronavirus disease-19 (COVID-19) through computational drug repurposing study. Journal of chemical information and modelling. 2020;60(6):3277-3286.
- [Google Scholar]
- WHO. (2019). “Middle East respiratory syndrome coronavirus (MERS-CoV).”.
- Zhai, P., Y. Ding, X. Wu, J. Long, Y. Zhong and Y. Li (2020). “The epidemiology, diagnosis and treatment of COVID-19.” International journal of antimicrobial agents55(5): 105955.
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2022.102214.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




