

Translate this page into:
A reaction of stabilized arsonium ylide of dicarboxylate ester with tropylium fluoroborate: An efficient strategy for ring contraction of cycloheptatriene
⁎Corresponding author. abdulmajeedsalihhamad@yahoo.com (Abdulmajeed S.H. Alsamarrai)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
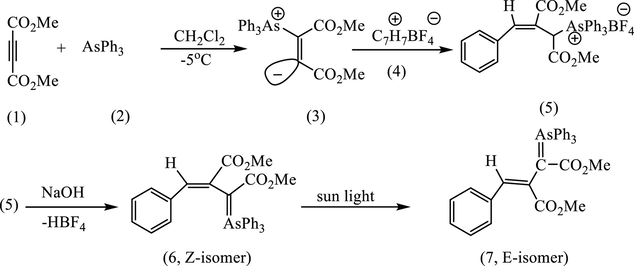
The coupling of vinylic ylide (3) synthesized from AsPh3 (2) and dimethyl acetylenedicarboxylate (DMAD) (1) with tropylium fluoroborate (4) in CH2Cl2 was found to give 3-phenyl-1,2-dicarbomethoxyprop-2-enylidentriphenyl arsonium salt (5) via ring contraction of cycloheptatriene. Treatment of the latter (5) with aqueous sodium hydroxide gives 3-phenyl-1,2-dicarbomethoxyprop-2-enylidentriphenylarsorane (6, Z-isomer), and this efficiently isomerized to E-isomer (7) under sunlight irradiation.
Keywords
Arsonium ylides
Vinylic ylides
Acetylenedicarboxylate
Triphenylarsine
Cycloheptatriene

1 Introduction
The cycloheptatriene ring is probably one of the most important structural components among a wide range of unsaturated carbocyclic compounds. This unsaturated ring is present in natural and unnatural compounds such as chamazulene (Ramadan et al., 2006), matricin (Ramadan et al., 2006), and azulene (Braun and Buechi, 1976; Wender and Filosa, 1976; Moore, 1977). One of the well-known aspects of the chemistry of cycloheptatriene is ring contraction. The contraction of the cycloheptatriene ring leads to the formation of the corresponding aromatic compounds generally considered to proceed via prior conversion to the norbornadiene valence tautomer (Reisman et al, 2011; Minegishi et al, 2003; McNamara and Maguire, 2011). The irreversible conversion of the latter to benzenoid products is greatly encouraged by the presence of appropriately situated leaving groups to see, for example, Scheme 1a, b. Numerous examples of type (a) ring contractions are found particularly among reactions of tropone and tropolone derivatives with nucleophilic reagents. Examples of type a and b (Scheme 1) are also reported in oxidation reactions of tropylium salts by various reagents, e.g., chromic acid (Scheme 1c) (Ghosh et al, 2013; Ghosh et al, 2014; Sar et al., 2016a, 2016b).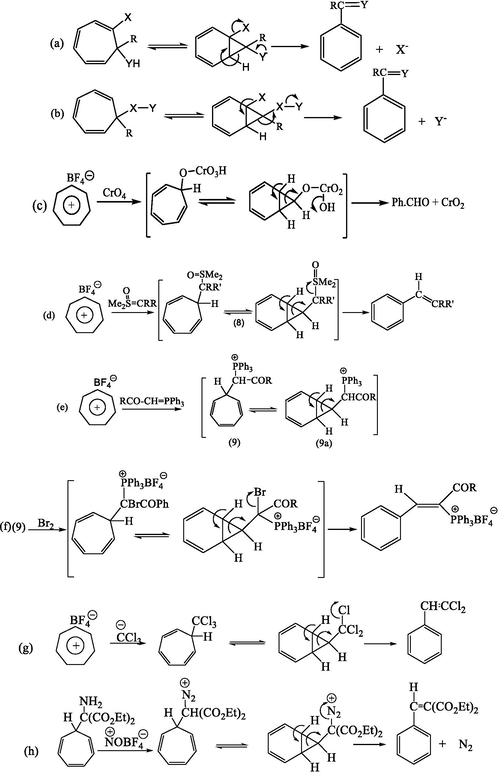
Examples of ring contractions of substituted cycloheptatriene.
Other examples, more relevant to the present work have recently been reported involving sulphoxonium (Cavicchio et al, 1977) and phosphonium intermediates (8) and (9) (Richards and Tebby, 1971), derived by the reaction of ylides with tropylium cation (Scheme 1d,e,f). In the case of the phosphonium intermediate (9), unlike other onium salts, the ring contraction (Cavicchio et al, 1980) did not take place spontaneously at room temperature with the expulsion of the onium group, and it is possible to isolate several salts of this structure. However, the introduction of an α-bromo atom which presumably has better properties as a leaving group resulted in the expected ring-contraction Scheme 1f. Other examples of this type of ring-contraction (Kohen and Weininger, 1972), can be seen in the Scheme 1g,h.
Thus, our continued interest in the chemistry of substituted cycloheptatriene compounds, derived from tropylium fluoroborate and nucleophiles encouraged us to investigate the reaction of vinylic ylides with tropylium fluoroborate. This process of reacting tropylium fluoroborate with vinylic ylide leads to the formation of substituted styrene bearing phosphonium moiety which could be exploited in a further reaction such as Wittig reaction to synthesize substituted dienes. The structural assignments of the new compounds were based on their IR, 1H NMR, 13C NMR, and UV data.
2 Materials and methods
Melting points were determined on Buchi melting point apparatus using an open capillary tube and they were uncorrected. All the required chemicals used were purchased from Aldrich without further purification. Acetonitrile used is of HPLC grade. Thin layer chromatography (TLC) was carried out on already made 5x25 cm plates coated with alumina oxide obtained from Merck and 5x25 cm plates coated with silica gel N-HR/UV245. The 1H NMR spectra were recorded on Brucker AVIII-HD-800 BioSpin 400 MHz and Varian Associates XL-100 instruments, while 13C NMR spectra were recorded on Brucher Shield 300 MHz by using TMS as internal reference and CDCl3 and DMSO‑d6. Chemical shifts (δ) are expressed in ppm relative to TMS as an internal standard on δ the scale. IR spectra were recorded on Shimadzu FT-IR-8400S and SP1050 spectrophotometers over a frequency range 4000–400 cm−1. Analytical high-pressure liquid chromatography was carried out using a cost metric III pump model 1204A connected to Spectro Monitor III variable. A reversed-phase column packed with 10 µm silica (partisil-10), 250 mm × 5 mm i.d. and spherisorb 5-ODS. The mobile phase composition of solvents CH2Cl2/MeOH/H2O is 95:4:1 by volume; the detector, 268 nm.
2.1 Reaction of triphenylarsine (2) with tropylium tetrafluoroborate (1) and Dimethylacetylenedicarboxylate(1) in
2.1.1 Dichloromethane solution
A suspension of tropylium tetrafluoroborate (1 g, 0.056 mol) in CH2Cl2 (10 ml) containing dimethyl acetylenedicarboxylate (0.8 g, 0.056 mol) was stirred under N2 at −5°C. Triphenylarsine (1.72 g, 0.056) in CH2Cl2 (10 ml) added dropwise and stirring was continued for 1 h after the addition was complete. The solvent removed in vacuo and the resulting white reside, m. p., 148–153 °C passed down a silica column using CH2Cl2: EtOAc (1:3) as eluent. The solvent evaporated and the yellow residue triturated with ether to give a yellow crystalline product (Yield: 2.1 g, 63%). HPLC of this material on Partisil-10 using CH2Cl2/MeOH/H2O (95:4:1) (detector 268 nm) showed the presence of two components. Three recrystallizations of a sample from the mixture of benzene and methanol afforded yellow crystals of 3-phenyl-1,2-dicarbomethoxyprop-2-enyltripnenylarsorane (7, Z-isomer) m.p. 172-174°, shown to be pure by HPLC (Found: C, 69.08; H, 5.20 C31H27O4As calculated C, 69.22; H, 5.11%). λmax (EtOH): 298 nm; υmax(KBr): 3100(CH aromatic), 3062 (CH olefinic); 2920 (CH-aliphatic.); 1730, 1692(C = O); 1625 (C = C aromatic). 1H NMR (DMSO‑d6): δ 7.55 (20H, m, C6H5); 6.40 (1H, s, H3); 3.60, 3.50 ppm (6H, 2xs, OCH3). A sample (100 mg) was dissolved in CH2Cl2 and stirred vigorously at room temperature, while 42% HBF4 solution was added until the yellow color completely discharged; the mixture stirred for a further 5 min. The organic layer was separated and washed with water and dried over anhydrous MgSO4. The CH2Cl2 solution concentrated and ether added until turbidity resulted and this was then discharged by the addition of little CH2Cl2. The resulting white product, which separated on keeping, was collected and washed with ether and dried giving crystals, m. p. 148-150°. Recrystallization of this sample from ethanol the solution turned yellow and on cooling to room temperature, this color discharged, and the resulting white crystals were collected and dried to give 3-phenyl-1,2-dicarbomethoxyprop-2-enyltripnenylarsiniumtetrafluoroborate of (7), m. p. 150-151°(Found: C, 59.20; H, 4.20 C31H28BF4O4As calculated C, 59.28; H, 4.45%).λmax (EtOH): 294 nm; υmax(KBr): 3106(CH aromatic), 3068 (CH olefinic); 2923 (CH-aliphatic.); 1734, 1697 (C = O); 1630 cm−1(C = C aromatic). 1H NMR (DMSO‑d6): δ 7.70–7.45 (20H, m, aromatic protons); 6.37 (1H, s, H3); 3.65, 3.54 ppm (6H, 2xs, OCH3); 3.10 ppm (1H, s, >CHAs+Ph3) see figure (1a).13C NMR(DMSO‑d6): general spectrum: δ 165.9, 165.2, 147.8, 133.2, 132.2, 131.7, 129.7, 128.4, 128.3, 121.9, 120.3, 53.1, 52.2, and 47.5; DEPT-90, δ 173, 152, 137.9, 137.4, 136, 135.4, 134.7, 127.4, 58.6, 57.7, and 52.9; DEPT-135, δ 152, 138.6, 137.7, 137.4, 137, 135.4, 134.8, 133.9, 133.7, 127.4, 58.6, 57.7, and 52.9 ppm.
2.1.2 Wet dichloromethane solution
The experiment above repeated in wet CH2Cl2 and the mixture stirred at −5°C for 3 h after the addition of AsPh3 was complete. A little insoluble material was filtered off and identified as tropylium fluoroborate (0.3 g. 30%) m. p. 210 °C (decomp.). The liquor mother evaporated in vacuo and treatment of the residue with ethyl acetate resulted in the separation of compound (6) identified as above. The solution was passed down a column of activated alumina using ethyl acetate as the eluent; the first component to be eluted recrystallized from ethyl acetate to give dimethyl fumarate, m. p. 97–98 °C [Lit., 102 °C] identified by comparison (IR and mixed m. p.) with an authentic sample (Yield: 250 mg 29%). Further elution gave As(O)Ph3, m. p. 188-190° [Lit., 191–192 °C] (Yield: 1.2 g, 67.4%).
2.1.3 Acetonitrile solution
The experiment in a repeated in dry acetonitrile (10 ml) and after completion of the reaction and evaporation of the solvent, compound (6) was isolated, purified, and identified as mentioned above (Yield: 2.6 g, 74%).
2.2 Preparation of the betaine (3a)
The betaine (3a) prepared as previously reported by Johnson and Tebby (1961). A solution of triphenylarsine (2 g, 0.0065 mol) in dry ether (30 ml) stirred and cooled to −50 °C in dry ice/ethanol bath; a rapid stream of CO2 passed through the solution via a sintered glass dispenser and a solution of dimethyl acetylenedicarboxylate (1.08 g, 0.0065 mol) in ether from a syringe was added dropwise during 20 min. The mixture was allowed to warm to 0 °C and the precipitate collected by suction and washed with ether, a little cold acetone and again with ether; it was dried at room temperature in vacuo, and used in the same day (Yield: 2.8 g, 87%).
2.3 Synthesis of 3-phenyl-1,2-dicarbomethoxyprop-2-enyltripnenylarsonium fluoroborate (5)
A suspension of the above betaine (3a) (2 g, 0.004 mol) in ether (20 ml) was stirred magnetically and cooled in ice; a solution of tropylium fluoroborate (0.76 g. 0.004 mol) in acetonitrile (20 ml) added and stirring continued at 0° for 1 h. After being allowed to warm to room temperature during a further 1 h, it finally refluxed for 20 min, the solvent removed in vacuo, the residue was taken up in CH2Cl2 (20 ml). Ether added until persistent turbidity resulted and this was then discharged by the addition of a little more CH2Cl2. The resulting pale cream crystalline product which separated on keeping was collected and washed with ether (Yield: 2.2 g, 75%). Recrystallization of sample from mixture of dichloromethane-ether gave the white crystalline product fluoroborate (5) (0.6 g, 60%) m.p. 149–151 °C (Found: C, 59.22; H, 4.30 C31H28BF4O4As calculated C, 59.28; H, 4.45%), λmax (EtOH): 300 nm, υmax(KBr): 3110(CH aromatic), 3070 (CH olefinic); 2925 (CH-aliphatic.); 1736, 1695 (C = O); 1632 (C = C aromatic), 1H NMR (DMSO‑d6): δ 7.67–7.42 (20H, m, aromatic protons); 6.40 (1H, s, H3); 3.60, 3.52 ppm (6H, 2xs, OCH3); 3.12 ppm (1H, s, >CHAs+Ph3). A sample of the prepared compound above dissolved in CH2Cl2 and a dilute solution of NaOH was added to the mixture and stirred vigorously for 5 min. The organic layer was washed with water and then evaporated. The yellow solid residue obtained was recrystallized twice from methanol to give crystals of (6) m. p. 162-163°. The compound was shown to be pure by HPLC using the conditions already described (Found: C, 69.02; H, 5.12 C31H27O4As calculated C, 69.23; H, 5.16%). λmax (EtOH): 340 nm; υmax(KBr): 3090 (CH aromatic), 3070 (CH olefinic); 2925 (CH-aliphatic); 1725, 1686 cm−1 (C⚌O); 1622 (C = C aromatic). 1H NMR (DMSO‑d6): δ 7.75 (20H, m, aromatic protons); 6.45 (1H, s, H3); 3.62, 3.53 ppm (6H, 2xs, OCH3).
2.4 Irradiation of 3-phenyl-1,2-dimethoxycarbonylprop-2-enyltriphenylarsonare (6, Z-isomer)
A sample of the ylide (6, Z-isomer) (100 mg) in CH2Cl2 (10 ml) in a Pyrex tube irradiated for 10 h using a tungsten lamp (300 W) positioned 10 cm from the tube. The reaction was followed by HPLC which indicated that the isomerization of the (6, Z-isomer) to the ylide (7, E-isomer) was complete after 10 h. Isolation of the product and comparison of its m. p. and IR spectrum with those of the (6, Z-isomer) confirmed their identity.
2.5 Reaction of 3-phenyl-1,2-dimethoxycarbonylprop-2-enyltriphenylarsonare (7, E-isomer) with tropylium fluoroborate
3-Phenyl-1,2-dimethoxycarbonylprop-2-enyltriphenylarsonare (0.7 g. 0.0013 mol) dissolved in CH2Cl2 (20 ml), finely powdered tropylium fluoroborate (0.23 g, 0.0013 mol) was added and the mixture stirred at room temperature until the color was discharged. The solvent was evaporated in vacuo and the white oily residue was triturated with ether collected and washed with ether giving dimethyl 1-(7-cyclohepta-1,3,5-trienyl)-1-triphenylarsono-3-phenylprop-2-ene-1,2-dicarboxylate fluoroborate (21) m. p. 163–166 °C (Yield: 0.7 g) (Found: C, 63.30; H, 4.60 C38H34BF4O4As calculated C, 63.68; H, 4.74%); λmax (EtOH): 272, 296, and 346 nm; υmax(KBr): 3020 (CH aromatic), 3073 (CH olefinic); 2990–2830 (CH-aliphatic); 1740 (C = O); 1590 cm−1 (C = C). 1H NMR (DMSO‑d6): δ 8.06–7.15 (20H, m, C6H5); 6.80–6.70 (2H, t; J2,3 3 Hz; H3 and H4 of 7-membered ring), 6.50 (1H, m), 6.30 (1H, s, PhCH = ), 6.18 (1H, m), 5.70–5.45 (1H, m); and 5.0 (1H, m) (olefinic protons of 7-membered ring), 4.50–4-35 (1H, m, H7) ; 3.50 and 3.30 ppm (6H, 2xs, OCH3).
2.6 Reaction of 1-(7-cyclohepta-1,3,5-trienyl)-1-triphenylarsono-3-phenylprop-2-ene-1,2-dicarboxylate fluoroborate (21) with sodium hydroxide in two-phase system
The fluoroborate above (21) (0.4 g, 0.0056 mol) was dissolved in CH2Cl2 (10 ml). 2 M Sodium hydroxide (10 ml), added and the solution vigorously stirred at room temperature for 2 h. The organic layer was separated, washed several times with water and dried over anhydrous MgSO4. The solvent removed in vacuo and the yellow oily residue was chromatographed on activated alumina using ethyl acetate as an eluent; dimethyl 1-(7-cyclohepta-1,3,5-trienyl)-3-phenylprop-2-ene-1,2-dicarboxylate (22) obtained as a colorless gum (140 mg, 77.7%). υmax (liquid film): 3020–2910 (C–H); 1750 (s), 1660 (s) (C⚌O), and1620, 1610 (w) cm−1 (C⚌C); λmax (n-pentane) 300 nm; 1H NMR (CDCl3): δ 7.5–7.15 (5H, m, C6H5), 6.83 (1H, s, vinylic proton), 6.68 (2H, t, H3 and H4 of 7-membered ring), 6.30–6.0 (2H, m; J2,3 3 Hz, J2,4 = J3,5 1.5; H2 and H5 of 7-membered ring), and 5.0 (2H, m; J1,2 7.5 Hz, J1,7 5.0 Hz, H1 and H6 of the 7- membered ring), 3.80 (3H, s, OCH3), 3.55 (3H, s, OCH3) , and 2.50 (1H, d; J1,(7) 5 Hz, H1 of the side chain), 2.30 ppm (1H, m, H7).
3 Results and discussion
The scope and usefulness of vinylidenes such as (10) (Scheme 2) as synthetic agents have little exploited. Apart from the work of Johnson and Tebby (1961) on the elucidation of structures of the various products obtained by generation of vinylidene (10) in the presence of the excess of one or other of the reagents, the literature appears to contain many references. One of these (Winterfeldt and Dillinger, 1966) describes the formation of lactone (10) and the benzoyl compound (11) in the reaction involving the slow addition of DMAD to a mixture of triphenylphosphine and benzaldehyde see Scheme 2.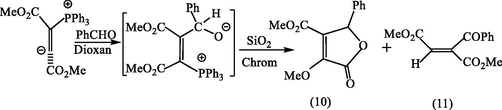
Reaction of phosphorus vinylic ylide with benzaldehyde in dioxan.
In another study (Burgada et al, 1980), the addition of trivalent phosphorus compound to DMAD, in the presence of various other trapping agents such as alcohols, has been reported. The general character of these reactions arises from the prior formation of the vinylidene, which then abstracts a proton from the donor.
3.1 The reaction of triphenylarsine (AsPh3) with dimethyl acetylenedicarboxylate (DMAD)
The present study describes the reaction of AsPh3 with DMAD in dry CH2Cl2 under nitrogen atmosphere and in the presence of an equivalent amount of tropylium fluoroborate, gives the adduct (5) m.p. 148–153 °C (Scheme 3). After recrystallization from a mixture of CH2Cl2: EtOAc (1:3), this recrystallization was accompanied by pale yellow color disappeared on cooling to room temperature. For further purification, the adduct chromatographed on silica gel, from which pale yellow compound (63%) eluted. After several crystallized from a mixture of benzene and methanol stood pale yellow needles, m.p. 172–174 °C of (7, E-isomer) shown to be pure by HPLC. These reactions clearly require the prior formation of the vinylic ylide (3), and by introducing carbon dioxide into the mixture of AsPh3 and DMAD, we were able to trap this intermediate as the carboxylate (3a) which was selectivity stable at <0 °C. This pale yellow product which supposed to be (5), however, evidently not arsonium salt, although it was readily converted into colorless salt by aqueous acids suggesting the presence of a stabilized ylide function.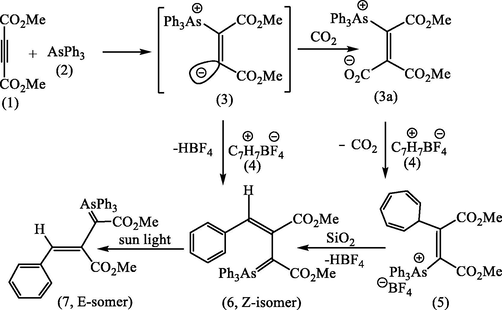
Syntheses of the isomers (6, Z-isomer) and (7, E-isomer).
The 1H NMR spectrum showed signals due to aromatic protons at δ 7.70–7.45 ppm (20H) and PhCH = signed at δ 6.37 ppm, and a pattern of 2 separate singlets at δ 3.65–3.57 ppm due to methoxy protons consistent with the presence of one isomer. However, the sample appeared to contain a single compound as shown by TLC, HPLC and its m. p. 162–164 °C could not be raised by further crystallization. Loss of HBF4 from (5) gave the ylide (6) which would be expected to occur readily in such a molecule, e.g., on silica gel chromatography, during the work-up. The 1H NMR of the white fluoroborate of (7) (Fig. 1a) also contained a one-proton singlet at δ 3.10 ppm clearly not coupled to another H-atom, this was missing in the NMR of the pale yellow compound (6). 13C NMR spectra (Fig. 1b-d) showed signals at δ 165.9 and 165.2 for carbonyl groups, 140–120 for aromatic and olefinic carbons, 53.1 ppm and 52.2 ppm for two OCH3 groups as well as a signal for C1 of >CHAs+Ph3 moiety.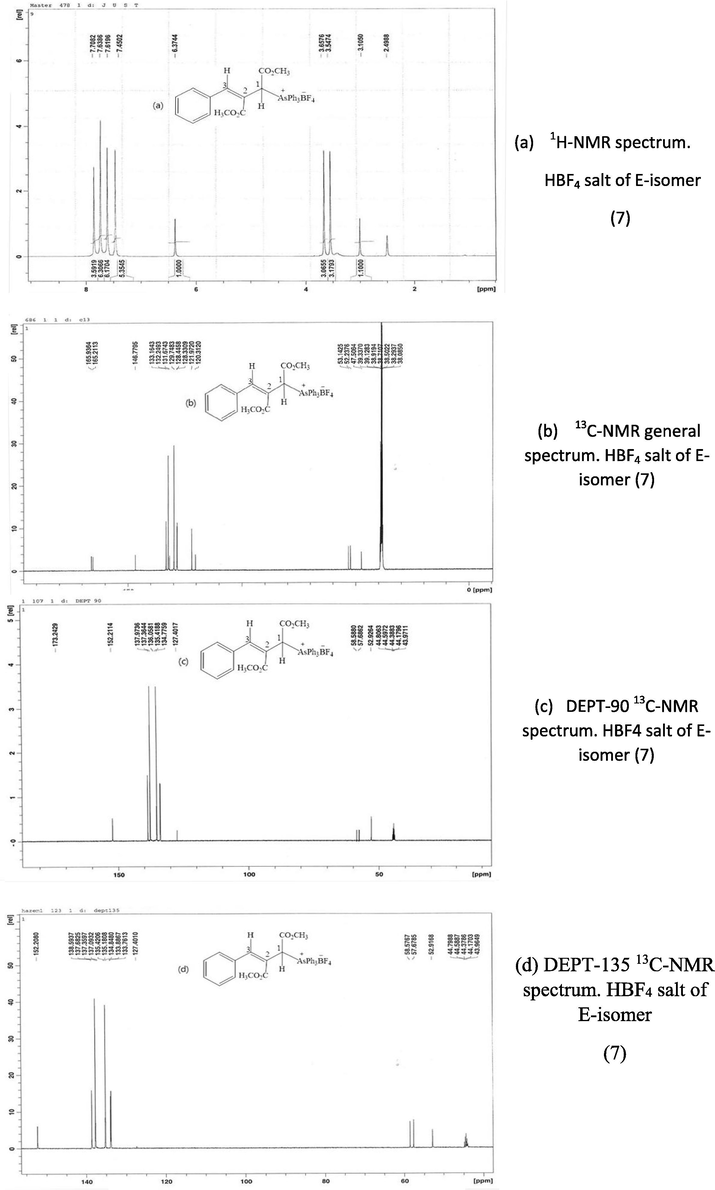
1H NMR spectrum. HBF4 salt of E-isomer (7) (b) 13C NMR general spectrum. HBF4 salt of E-isomer (7) (c) DEPT-90 13C NMR spectrum. HBF4 salt of E-isomer (7) (d) DEPT-135 13C NMR spectrum. HBF4 salt of E-isomer (7).
It was then observed that the reaction of DMAD and AsPh3 was very susceptible to change insolvent. In dry dichloromethane, the adduct (3) had only obtained, but in ethanol, chromatography of the ethanol-soluble products on silica gel afforded only dimethyl fumarate. The latter reported (Richards et al, 1969) to result from the reduction of DMAD and TPP in refluxing aqueous THF for several hours, evidently as a result of Michael addition followed by hydrolysis at phosphorus see Scheme 4.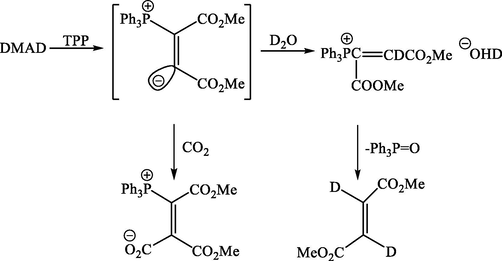
Hydrolysis of phosphorus vinylic ylide by D2O.
In the absence of protic solvents, it previously reported that the vinylic phosphorus ylide combines with unreacted DMAD and or TPP to give various adducts, the structures (12–14) of which have been elucidated by Johnson and Tebby (1961).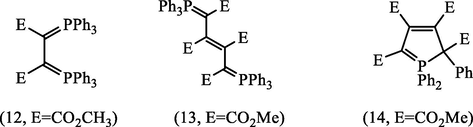
In aprotic media (CH2Cl2, CH3CN) as it has been mentioned earlier in this study, the vinylidene (3) would be expected under these conditions to quench the tropylium cation (4) to give (5). However, again, neither the 1H NMR spectrum of the yellow ylide, nor that of its HBF4 (Fig. 1a) salt were consistent with this structure in particular in the absence of signals expected for the six protons of a cyclic triene, and consistent with the ylide (6) which formed by loss of HBF4 during the silica chromatography see Scheme 3. Various attempts to improve the yield, e.g., one of them by combining the reagents in a different order. Addition of AsPh3 in CH2Cl2 to DMAD and tropylium fluoroborate in the same solvent did indeed afford improved yields of the yellow ylides (6 and 7). An even more effective route to this product was developed by reacting the carboxylated ylide (3a) directly with tropylium fluoroborate in acetonitrile. This gave directly a colorless fluoroborate (5) m.p. 150–151 °C in 74% yield, which on basification and isolation with CH2Cl2 gave a yellow crystalline ylide (6), see Scheme 5. Regeneration of the fluoroborate of (6), has been achieved by treating the ylide (6) with aqueous HBF4. However, we found that careful analysis by HPLC (50 cm × 5 mm Partisil 10 using 95:14:1 CH2C12/MeOH/H2O; detector, 268 nm) of crude samples of the ylide (6), obtained earlier, showed the presence of second minor component, which proved to be identical with the compound synthesized as described above. It was subsequently shown by using analytical HPLC that the second ylide, m.p. 162–163 °C could be isomerized to the first by irradiation of CH2C12 solution with the 300w tungsten lamp. This indicated that their relationship was that of a pair of geometrical isomers and suggested that the higher-melting ylide (172–174 °C) had been obtained originally as a result of inadvertent photoisomerization during the work-up, probably in the course of chromatography on silica on a sunlit bench. The structural problems presented by the spectroscopic properties of the two ylides (6 and 7) and their conjugate acids and their geometrical isomerisms were resolved by realization that the 7-membered ring of the initial product (5) has undergone ring-contraction at some stage in the reaction see Scheme 3.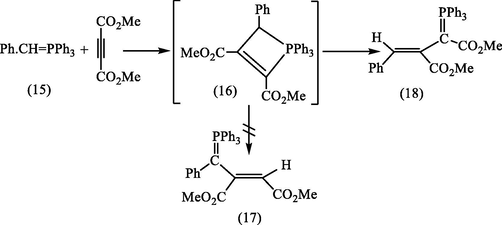
Bestman and Rothe protocol for the synthesis of the ylide (17).
3.2 The ring contraction in compound (5)
In the present case, therefore, ring contraction can be understood in terms of the norcaradiene structure (5a), in which cleavage of the cyclopropane ring is facilitated by the great stability of the resulting ylide (7) during the chromatography elution on silica gel.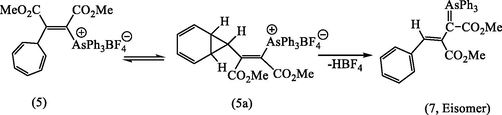
It was shown by Bestmann and Rothe (1964) that the reaction of DMAD with the ylide (15), which was previously reported by Trippett (1962) to give (17), instead gave the same conjugated ylide (18), via a 4-membered ring intermediate (16) (Scheme 5). Bestmann and Rothe's compound was shown by X-ray crystallography (Lingner and Burzlaff, 1974) to have the geometry depicted in (17) with the phenyl and CO2Me groups in cis-relationship.
The formation of a single geometrical isomer (7, E-isomer), in our reaction presumably arises through minimization of steric compression in the opening 4-membered ring in the intermediate (19) (Scheme 6).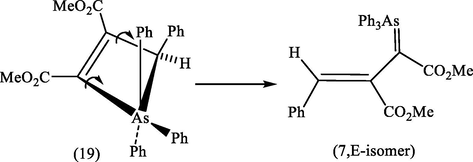
4-Membered ring intermediate (18) collapses to (7-E-isomer).
Hence the compound (7, E-isomer) formed by the addition of the vinylide (3) to tropylium fluoroborate should have the stereochemistry is shown in which case the bulky triphenyl arsonium group would be expected to result in a preference for confirmation (5b) rather than (5a) (Scheme 7). Cleavage of the cyclopropane ring in such a confirmation would naturally lead to the establishment of the new phenyl group cis to the CO2Me rather than to the ylide function, as in the structure (7, E-isomer).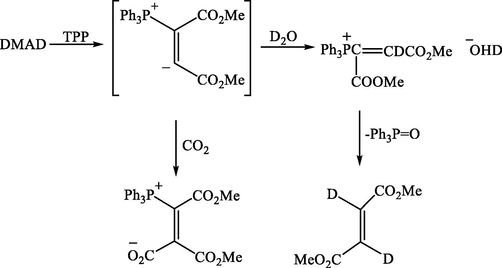
Syntheseis of dimethyl fumarate via vinylic ylide.
Recently, few of these (Gao et al., 2013) describe the formation of vinylic ammonium ylides used in the formation of functionalized 2-pyrrolidine-3-olates, pyrans and polysubstituted benzene (Moustafa et al., 2014). Another recent study has demonstrated the reaction of hydrazines and acetylene dicarboxylate in the synthesis of functionalized 1-benzamidospiro [indole-3,4-pyridines] (Wang et al., 2014).
3.3 The reaction of the ylide (7, E-isomer) with tropylium fluoroborate
It seems that the ylide (7, E-isomer) or even the ylide (6, Z-isomer) are potentially reactive, it, therefore, seemed reasonable to examine the reactivity of the ylide (7, E-isomer) towards tropylium fluoroborate. The pale yellow ylide (7, E-isomer) was reacted with an equimolar proportion of tropylium fluoroborate in CH2Cl2 at room temperature. Decolorisation of the pale yellow ylide occurred instantly, and a white crystalline product, m.p. 163–166 °C was obtained on evaporation of the solvent which was insufficiently stable to withstand further recrystallization from organic solvents. The 1H- NMR spectrum of this product showed several multiplets between δ 6.80 and 4.90 and integrating for six olefinic protons and a vinylic proton H10. In addition, a one proton multiplet appeared at δ 4.35 attributable to the 7-proton of a monosubstituted cycloheptatriene, and consistent with structure (20).In the present case, unfortunately, the NMR spectrum did not show such a distinguished triplet for the coupling of H7 with H1 an H6 which seemed to be partially hidden under one of the methoxy group absorptions centered at δ 3.40 ppm.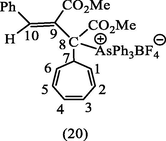
An attempt to purify the arsonium salt (20) by recrystallization from ethanol failed, and the solution became yellow, suggesting the reversion to the ylide (7, E-isomer). After allowing the solution to cool down, the yellow color discharged and impure crystals of (19), m. p. 162–170 °C were again obtained, suggesting that the arsonium salt (20) was in equilibrium with tropylium fluoroborate and the ylide (7), dissociation being favored in hot solvent. The absence of ring contraction of the cycloheptatriene ring in this case can be attributed to the fact that the + AsPh3 group is a poor leaving group and is in an inappropriate position to stabilize the negative charge arising from cleavage of the cyclopropane ring of the norcaradiene valence isomer (21), in contrast to the situation in (5b). However, the treatment of the arsonium salt (20) in dichloromethane with 2 M-sodium hydroxide at room temperature gave a white gummy product which was eluted from an activated alumina column using ether as eluent. Spectroscopic data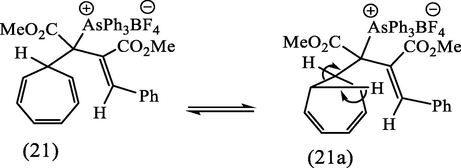
and elemental analyses indicated that the triphenyl arsonium residue had been eliminated from this product, and the IR spectrum showed that both saturated and unsaturated ester groups were present. The 1H NMR spectrum exhibited three multiplets centered at δ 6.7, 6.15, and 5.1, typical of a 7-substituted cycloheptatriene, and also a multiplet centered at δ 2.30 attributable to the 7-proton coupling with H1 and H6 of the cycloheptatriene ring as well as coupling with an adjacent proton in the side chain. This proton appeared as a doublet centered at δ 2.50 ppm (J 5.5 Hz). These data, together with the identification (TLC) of As(O)Ph3 in the hydrolysis products, suggested that the structure of this product was the 7-substituted cycloheptatriene (22), formed according to Scheme 8.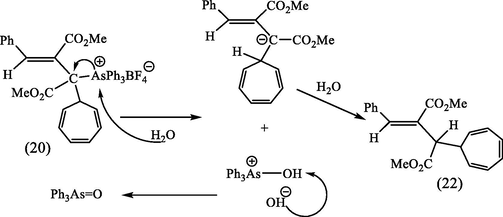
Reaction of the compound (20) with aqueous NaOH.
4 Conclusion
In conclusion, we have demonstrated a further example of ring contraction of cycloheptatriene accelerated by the presence of arsonium group in position-7. This operationally simple protocol opens the route to the synthesis of E-isomers of 3-phenyl-1,2-dicarbomethoxyprop-2-enyltripnenylarsorane. Despite the synthetic utility of the vinylic ylides presented herein, the field of reactions involving these versatile building blocks remains insufficiently explored. It is likely that this ring contraction under this mild condition is effective from the viewpoint of synthetic chemistry, as an introduction of functionalized alkenes in good yields with high purity.
Acknowledgment
Financial support from the college of education at Samarra university is greatly acknowledged. We also thank Mr. Bakir Ismael for his preliminary technical assistance.
References
- The reaction of Alkylidenetriphenylphosphoranes with acetylenedicarboxylic esters. Angew. Chem. Int. Ed.. 1964;3:512-513.
- [Google Scholar]
- Trapping of carbanionic species synthesis of new salt-free ylids and phosphoranes. Tetrahedron Lett.. 1980;21:925-928.
- [Google Scholar]
- The reaction of stabilized ylides and related onium salts with tropylium ion. Tetrahedron Lett.. 1977;18:3493-3496.
- [Google Scholar]
- Synthesis of (2, 4, 6-cycloheptatriene-1-ylidene) methyl triphenylphosphonium tetrafluoroborate: a strongly stabilized ylide. Tetrahedron Lett.. 1980;21:2333-2336.
- [Google Scholar]
- A Four-component reaction of cyclic amines, 2-aminobenzothiazole, an aromatic aldehyde, and acetylenedicarboxylate. Beilstein J. Org. Chem.. 2013;9:2846-2851.
- [Google Scholar]
- A suitable combination of promoter and micellar catalyst for kilo fold rate acceleration on propanol to propionaldehyde conversion in aqueous media. J. Ind. Eng. Chem.. 2014;20:345-355.
- [Google Scholar]
- A suitable combination of promoter and micellar catalyst for kilo fold rate acceleration on benzaldehyde to benzoic acid conversion in aqueous media at room temperature: A kinetic approach. Spectrochim. Acta A. Mol. Biomol. Spectrosc.. 2013;109:55-67.
- [Google Scholar]
- The adducts from triphenylphosphine and dimethyl acetylenedicarboxylate. J. Chem. Soc. 1961:2126-2130.
- [Google Scholar]
- Die Kristall-und molekülstruktur des 1, 2-Bis (methoxycarbonyl)-3-phenylpropene-2-ylide-1-triphenylphosphine. Acta Crystallogr. Sec. B. 1974;30:1715-1722.
- [Google Scholar]
- Acid-Catalyzed conversion of 7-ethynyl-and 7-vinylcyclohepta-1,3,5-trienes to substituted benzene derivatives. Eur. J. Org. Chem.. 2003;2003(18):3497-3504.
- [Google Scholar]
- Organic bases-catalyzed three-component reactions for the synthesis of 4H-2-aminopyrine, condensed pyrans, and polysubstituted benzenes. Beilstein. J. Org. Chem.. 2014;10:141-149.
- [Google Scholar]
- Chamazulene carboxylic acid and matricin: a natural protein and its natural prodrug, identified through similarity to synthetic drug substances. J. Nat. Prod.. 2006;69:1041-1045.
- [Google Scholar]
- Buchner and beyond: Arene cyclopropanation as applied to natural product total synthesis. Synlett. 2011;2011(17):2437-2442.
- [Google Scholar]
- Reactions of phosphines with acetylenes. Part XII. The mechanisms of 1, 2-aryl migrations common to αβ-unsaturated phosphonium salts and “Wittig-type” phosphonium betaines. J. Chem. Soc. (C) Org. 1971:1059-1063.
- [Google Scholar]
- Reactions of phosphines with acetylenes. Part VIII. Synthesis of 1, 2-dideuteriated olefins. J. Chem. Soc. (C): Org. 1969;11:1542-1544.
- [Google Scholar]
- The combined effect of promoter and surfactant on the chromium (VI) oxidation of D-ribose in aqueous media at room temperature. J. Carbohydr. Chem.. 2016;35:86-105.
- [Google Scholar]
- Selective heteroaromatic nitrogen base promoted chromium (VI) oxidation of isomeric pentanols in aqueous micellar media at room temperature. J. Ind. Eng. Chem.. 2016;42:53-62.
- [Google Scholar]
- Addition of Wittig reagents to activated carbon-carbon double bonds. J. Chem. Soc. 1962:4733-4734.
- [Google Scholar]
- One-pot four-component reaction for convenient synthesis of functionalized 1-benzamidospiro [indoline-3, 4'-pyridines] Beilstein. J. Org. Chem. 2014;10:2671-2676.
- [Google Scholar]
- A facile route to divinylcyclopropanes. An efficient method for the annelated formation of functionalized cycloheptanes. J. Org. Chem.. 1976;41:3490-3491.
- [Google Scholar]
- Additionen an die Dreifachbindung, VI. Heterocyclen aus Acetylenverbindungen. Chem. Ber.. 1966;99:1558-1568.
- [Google Scholar]




