

Translate this page into:
A novel homozygous splice site variant in the CLCN7 causes osteopetrosis
⁎Corresponding authors at: Chairman of Genetics and Precision Medicine Department (GPM), Director of Medical Genomic Research Lab, King Saud Bin Abdulaziz University for Health Sciences, King Abdulaziz Medical City, Ministry of National Guard Health Affairs (MNGHA), Riyadh, Saudi Arabia, P.O Box: 22490 Riyadh 11426 mail code 1490 (M. Alfadhel). zainab17@windowslive.com (Zeinab Klaab), dralfadhelm@gmail.com (Majid Alfadhel)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Objectives
Osteopetrosis is a monogenic disorder represented by disturbed osteoclast resorption or osteoclastogenesis differentiation. Clinical symptoms are intensive and brittle bones, recurrent fractures, thrombocytopenia, impaired immune function, optic nerve compression, and anemia. Several osteopetrosis-causing genes have been identified and reported.
Methods
The present study describes two consanguineous Saudi families segregating a severe autosomal recessive osteopetrosis disease. A single proband (II-2) in family A and two probands (II-2; II-4) in family B exhibited increased bone density, multiple fractures, teeth abnormalities, bilateral optic atrophy with nystagmus, and progressive blindness. DNA of the affected individuals was exposed to whole-exome sequencing (WES) and Sanger sequencing. Further, reverse transcriptase-polymerase chain reaction (RT-PCR) and western blotting analysis were done to investigate the impact of the identified mutation.
Results
WES revealed a novel homozygous splice site variant (c.739-18G > A) in the CLCN7 gene on chromosome 16p13.3, segregating perfectly with the disorder phenotype. RT-PCR resulted in the retention of a 50 bp sequence of intron 8 in the mutated sequence. As a result, this variant resulted in a large size exon 9 compared to the wild type. In addition, the western blot revealed the heteromeric form of ClC-7 disappeared in the patient’s fibroblasts versus the control, indicating identified variant pathogenicity.
Conclusion
The present research provides certain proof that homozygous variants in the CLCN7 gene are responsible for intense osteopetrosis disorders with diverse phenotypes. These findings have significant implications for decisions regarding the clinical therapeutic regimen, prognosis assessment, and antenatal diagnosis.
Keywords
CLCN7
Osteopetrosis
Osteoclast
Splice site variant
Autosomal recessive

1 Introduction
Osteopetrosis is a rare, genetic, heterogeneous, and potentially life-threatening disorder. The disease is distinguished as skeletal dysplasia; symptoms include increased bone density, decreased bone strength, brittleness, and stenosis of the medullary cavity (Khan et al., 2018; Penna et al., 2019; Bug et al., 2020).
Numerous types of osteopetrosis as autosomal recessive osteopetrosis (ARO) (OMIM #259700) have been recorded; sometimes termed a “malignant” and “infantile” form, autosomal dominant osteopetrosis (ADO) (OMIM #166600), often known as Albers-Schönberg or “benign” disorder, intermediate recessive autosomal osteopetrosis (IARO) of a less severe form (OMIM #259710) and X-linked osteopetrosis (XLO) (Drake et al., 2017; Li et al., 2019).
The prevalence of osteopetrosis is less common than reported to be 1:200,000. Still, it can be much higher (Amirfiroozy et al., 2017), whereas ADO is the most prevalent form of osteopetrosis, with an incidence of 1:20,000 (Khan et al., 2018). In addition, a few cases of XLO have recently been described (OL-EDA-ID syndrome). However, IARO remains the less prevalent type, with only a few patients found worldwide thus far (Del Fattore et al., 2008).
ARO malignant form is apparent in early infancy or childhood and has a poor prognosis. The patients present with pathological abnormalities in various systems, for instance: hepatosplenomegaly, pancytopenia, hematopoiesis failure, recurrent infections, hydrocephalus, and skeletal system deformities (Kuroyanagi et al., 2014). Furthermore, compression of the cranial nerves affects neurologic functions and causes nerve palsies, facial (7th) paralysis, progressive deafness, blindness, and swallowing problems (Palagano et al., 2017; Penna et al., 2019).
In addition, biochemical features such as increased serum levels of aspartate transaminase (AST), lactate dehydrogenase (LDH), tartrate-resistant acid phosphatase-5b (TRACP-5b), and the creatine kinase (CK) BB isoenzyme (CK-BB) may be beneficial in the prognosis of osteopetrosis (Pang et al., 2016).
Mutations cause osteopetrosis in at least ten genes that have been reported the defects in osteoclast function or differentiation in humans. They collectively represent around 80 % of patients in the various cohorts (Bug et al., 2020). The genetic basis for osteopetrosis disorder is currently well interpreted: mutations in CLCN7 (OMIM 602727), TCIRG1 (OMIM 604592), OSTM1 (OMIM 607649), PLEKHM1 (OMIM 611466), and SNX10 (OMIM 614780) genes cause osteoclast-rich ARO. While mutations in (TNFSF11, MIM 602642) and (TNFRSF11A, MIM 603499) genes cause osteoclast-poor ARO (Wu et al., 2017). Of these genes, the majority of ARO cases are caused by biallelic variations of OSTM1 (2 %–6%), CLCN7 (13–16 %), and TCIRG1 (50 %), respectively.
In the current study, a novel splice acceptor site variant in the CLCN7 gene in two families with autosomal recessive osteopetrosis was detected using whole-exome sequencing (WES).
2 Materials and methods
2.1 Blood mononuclear cells isolation and study approval
Peripheral blood mononuclear cells (PBMCs) were gathered from the probands (II-1) from family A; (II-2; II-4) from family B and their normal parents and siblings (Fig. 1). According to the manufacturer's recommendations, PBMCs were separated using Ficoll-prefilled Leucosep® tubes by density gradient centrifugation. The trypan blue exclusion test was used to determine the present viability of PBMC (cells should be > 85 %-90 % viable). The King Abdullah International Medical Research Center (KAIMRC), Ministry of National Guard, Health Affairs, Riyadh, Saudi Arabia, served as the source of patients for this study. The Institutional Review Board provided their approval to the present study (approval# KAIMRCIRB- H-01-R-005) according to Helsinki protocols. Written informed consents were obtained from the parents to publish these cases, photographs and their molecular data.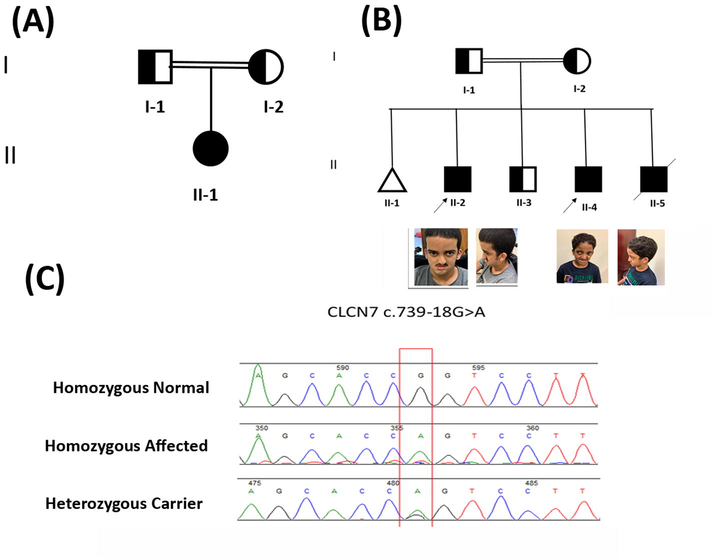
Pedigrees and sequence analysis of the families with autosomal recessive osteopetrosis. (A), (B) Pedigree of the osteopetrosis family A indicating affected family members (fully shaded). The arrow shows the probands. (B) Images of Probands with CLCN7 variant. Representative images of the probands at age 18 and 12 show the patient's face and ears, respectively. (C) Sanger sequencing chromatograms show the reference sequence in the top panel. The middle panel represents the nucleotide substitution in the patient's homozygous sequence. The last one represents the carrier individual in heterozygous states, respectively.
2.2 Genomic DNA extraction
High-quality DNA was extracted manually utilizing the QIAamp® DNA Mini kit method (QIAGEN®, Germany) from the whole blood according to the manufacturer's guidelines. Briefly, cellular membranes were ruptured by mechanical lysis and enzymatic activity (shaking and heating); DNA was bound to the silica-gel membrane of the QIAamp spin column; the impurities were washed across the membrane using buffers, and DNA was eluted. Quantification of gDNA (ng/μl) and purity (A260/A280) were assessed by a Nanodrop™ spectrophotometer (ND-1000, V3.8.1, NanoDrop Technologies, U.S.A.) following slandered methods.
2.3 Whole-exome sequencing (WES)
Whole-exome sequencing was achieved for each patient (II-1) from family A; (II-2; II-4) from family B described (Alfadhel et al., 2021a,b). The genomic DNA (g-DNA) was fractionated using a solicitor, and Illumina adapters were ligated and sequenced on the HiSeqX platform (Illumina) with an average ∼ 30X coverage depth. Further, an in-house pipeline base calling was performed. Diseases-causing variants were illustrated in ClinVar, HGMD®, CentoMD®, or PubMed and variants with minor allele frequency (MAF) of<1 % in the ExAC/gnomAD database were given preference.
All inheritance patterns were considered during the filtration of variants; however, homozygous and compound heterozygous variants of the pedigree recessive pattern were favored. The study focused primarily on pathogenic disease-causing non-synonymous (NS) variants. Moreover, clinical details and phenotypes provided from patients were used to demonstrate genotype-phenotype correlation.
2.4 In silico prediction
In silico prediction applications used to investigate the identified variant included MutPred Splice, NNSplice (Berkeley), Human Splice Finder, SKIPPY, MutationTaster, and Varsome.
2.5 Sanger sequencing
Sanger sequencing was performed on all the available family members to verify the results identified in the patients using WES. The primers for CLCN7 (NM_001287.5) were designed using Primer3 online software (https://frodo.wi.mit.edu/primer3/). The following primers sequence were used for PCR amplification forward primer 5‘-GGC ACA CTT GGT CTC AGG TT −3‘ and reverse primer 5‘- GTG CTG TTT TAC GGC TCA GG −3‘. The target sequences were amplified using PCR and then examined by agarose gel (2 %) electrophoresis. The sequencing was performed on a 3730 XI DNA Analyzer (Applied Biosystems®, HITACHI, Japan). The acquired sequence data were interpreted using Mutation Surveyor® software, version 5.1.2.
2.6 RNA extraction
Total RNA was extracted from the (1 × 108) PBMCs samples and cultured fibroblasts (106 cells) of patients using the QIAamp® RNA Mini kit method (QIAGEN®; Germany) following manufacturer instructions of blood and cells, respectively. Extracted RNA purity and quantified was assessed using a nanodrop™ spectrophotometer (ND-1000, V3.8.1, NanoDrop Technologies, U.S.A.) following standard procedures (Asiri et al., 2020).
2.7 Rt-PCR
To quantitatively monitor CLCN7 (NM 001287.5) mRNA expression to the internal control “house-keeping” gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH); DQ403057, total RNA was extracted, and the high-capacity cDNA reverse transcription kit (Applied Biosystems; Lithuania) was used to create the cDNA by PCR-recommended cycles in four steps: 25 °C for 7 min, 37 °C for 120 min, 85 °C for 5 min and at four °C for ∞. RT reactions were performed using 2 μg of RNA, previously subjected to DNase digestion and reverse transcription reagents, followed was subjected to thermocycler (applied biosystems®, Veriti; 96 Well Thermal Cycler, REF:4375786, Singapore). The primer sequences for the CLCN7 cDNA amplification and GAPDH will be shared upon request. The primers were designed using Online Primer3 software (https://frodo.mit.edu/primer3/).
The RT-PCR reaction was executed using an optimized Dream Taq Green PCR Master Mix (2X) (Thermo Scientific®, Lithuania) on a thermocycler PCR System (Applied Biosystems®, Singapore) by PCR recommended cycles. The initial denaturation step at 95 °C for 7 min; then 35 cycles at 95 °C for 30 sec (Denaturing); for 30 sec (Annealing) at the different temperatures depending on the primers that were used, and at 72 °C for 1 min (Extention); and a further extension at 72 °C for 7 min. All the RT-PCR reactions were carried out in triplicate and repeated independently. The results were analyzed on agarose gel (2 %) containing trace amounts (0.3 µg/ml) of ethidium bromide. The gel was run for 35 min, 120 V (BioRAD®, REF: 041BR 25589, USA) and visualized by ChemiDoc™ MP Imaging System with UV light (BioRAD®, REF: 731BR 03013, USA) (Asiri et al., 2020; Lien et al., 2022).
2.8 Protein extraction and western blotting
Whole-cell protein lysates of fibroblast cells (106 cells) were performed on 100 µl of lysis buffer 6 (biotechne®, USA) and 1 µl of protease inhibitors (Thermo Scientific, USA). Protein Quantification was performed using Qubit Fluorometer 3.0 (Invitrogen, REF: Q33216, USA). Protein was loaded in equal amounts (100 µg) into the wells of a Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) gel, along with a molecular weight marker (BIO-RAD, Singapore). Proteins were transferred to the PVDF membrane to complete the steps of western blotting. The western blot signals were measured using an ODYSSEY CLx (LI-COR®, Model: 9140, USA). The Actin antibody was used as a reference for loading. The antibodies used in the present study include Rabbit Polyclonal anti-ClC-7 Antibody, 1:2500 (Thermo Scientific, Cat. No. PA5-60203, USA). Mouse Polyclonal anti-Actin Antibody (loading control), 1:1000 (Abcam, Cat. No. ab1801, USA). Goat anti-Rabbit 926–68071, IRDye® 680RD, 1:10000 (LI-COR, Lot NO. C40723-01, USA). Goat anti-Mouse 926–32210, IRDye® 680RD, 1:10000 (LI-COR, Lot NO. C50113-06, USA).
2.9 Statistical analysis
One-way ANOVA test was utilized to interpret the functional study result, including Western blot; all findings with a P-value of < 0.05 were regarded as significant (Al Mutairi et al., 2020).
3 Results
3.1 Family A
3.1.1 Case description
A 4-year-old Saudi girl revealed features such as the dilated ascending aorta, bilateral abnormal middle ear function, nystagmus, and macrocephaly. She was referred to a genetic service because of a family history of osteopetrosis. The pregnancy course was unremarkable except for gestational diabetes mellitus (GDM). Delivery was at full-term (FT), spontaneous vaginally delivered (SVD). After delivery, she was transferred to the neonatal intensive care unit (NICU) and post exchange transfusion. She had polycythemia, hypocalcemia, thrombocytopenia, and hypomagnesemia; after that, she was discharged in good health. The family's first concern was at the age of 6 months when the mother observed abnormal eye movement (nystagmus) and increased the head's size. The patient is following well. No seizures. Family history includes consanguineous parents. They have another son and daughter; both are healthy.
Developmental history showed gross developmental delay. She (II-1) walked with support. The fine motor displayed by the index can through objects. Also, she could say baba and mama. The patient was socially active (Fig. 1a).
Physical examination revealed a weight of 9.4 kg [<1 percentile (-3.9 SD)] and a standing height: of 75.6 cm [<1 percentile (-6.0 SD)]. Head circumference (HC) was 48 cm [15 percentile (-1.04 SD)]. There was head and neck nystagmus retrognathia. A chest and abdomen examination of the patient was normal. Cardiovascular (CVS) examination perfused well. Audible heart sounds showed no added sounds. Gastrointestinal (GI) examination showed soft and lax and no organomegaly. The genitourinary examination was normal female. The index was not noticed any musculoskeletal (MSK) deformity. Neurological examination observed normal power, tone, and reflexes.
Skeletal X-rays showed generalized increased bone density involving the whole skeleton. Furthermore, a definite fracture was not identified. The index (II-1) 's overall appearance suggests osteopetrosis for clinical and genetic correlation (Fig. 1-A). Echo examination revealed dilated ascending aorta. The affected individual suffered from global developmental delay (GDD).
4 Family B
4.1 Case description
An affected individual is an 18-year-boy (II-2) with a history of multiple fractures, bilateral optic atrophy with nystagmus, right lower motor neuron (LMN), facial palsy, and a similar history in his brother (II-4), 12-year-boy-old, were investigated in the present study (Fig. 1B).
Medical history explained the first concern of the affected individual at 12 months when his mother was noticed delayed walking and decreased eyesight. After two years, he developed fractures. Pregnancy was unremarkable, and the birth was full-term normal spontaneous vaginal delivery (FT- NSVD). The index had only jaundice postnatally.
Developmental history showed gross standing and walking at three years. However, now the patient is restricted to a wheelchair due to femur (thigh bone) fractures. Furthermore, he had poor vision and used the braille style that reflected fine motor development. The proband could say single words at three years; however, now he is partly talking. The affected individual (II-2) is socially up to his age; however, he suffers developmental delay (Fig. 1 B). Family history includes healthy first-cousin parents; they have a healthy-one brother and two affected brothers. The first one had a fracture and unilateral blindness (II-4), but the other died at ten months with the same thrombocytopenia (II-1) (Fig. 1 B). Their paternal aunt, 35 years, had hydrocephalus with shunt and bone disease, multiple recurrent fractures, and osteogenesis.
Physical examination revealed his height and weight were 152 cm [< 1 percentile (-3.1 SD)] and 52 kg [5 percentile], respectively. His Head circumference (cm) was 59.5 [> 99 percentile (+3.1 SD)]. The index's head and neck dysmorphic showed dolichocephaly, long face, prognathism, high forehead, prominent nose, and low set ears. CVS examination reflected well perfused. GI examination explained no distended organomegaly. The affected individual suffered from vitiligo. Radiographs of musculoskeletal showed a general greater bone density with multiple fractures.
4.2 WES and Sanger sequencing
The two families (A and B) were subjected to WES using standard methods (Alfadhel, 2019; Alfadhel et al., 2021a,b). Step-by-step filtering and validation of various compound heterozygous and homozygous variants revealed a splice site variant (c.739-18G > A); (p.Met250Argfs*6) in the intron 8 of the CLCN7 gene (NM_001287.5; NP_001278.1).
Using the Sanger sequencing method, the identified variant segregated perfectly with the disease phenotypes in both families (Fig. 1 C). The homozygous state of the variant was present in the patients. To exclude the non-pathogenic of the identified variant, it was screened within ExAC, gnomAD, and 2000 Saudi exome databases. The pathogenicity index was calculated using various online analysis applications [(FATHMM-MKL: Damaging, Mutation Taster: Disease-causing, Varsome: PM2, PP3, DANN: 0.9924)] and was predicted disease-causing variant. The CLCN7 variant (c.739-18G > A) is novel and not reported in any currently genetic variant databases such as the human gene mutation database (HGMD).
4.3 Rt-PCR
To independently validate and quantify the splice defect identified by WES and Sanger sequencing, we performed an RT-PCR assay using primers flanking intron 8 from exon 7 to exon 9. Interestingly, RT-PCR results confirm significant retentions of intron 8 in patients' samples (Fig. 2 A). These findings expand the understanding of the association between a synonymous variant of CLCN7 and the molecular pathogenesis in osteopetrosis patients.![(A) Gel image showing the RT-PCR results of the index, father, mother and control samples. Lane (L: 1) showing 100 bp ladder, lane (L:2) index [449 bp], lane (L: 3) father, lane (L: 4) mother, and lane (L: 5) represented the control [399 bp] (healthy individual). The control shows a clear single band size of 399 bp. While the index showed a 449 bp band, suggesting the insertion of 50 bp in the index, both parents showed double bands of 449 bp and 399 bp. (B) Schematic representation of CLCN7 exons and introns. Arrow represents the identified variant's position and retention of the 50 bp sequence of intron 8. As a result, this variant results in large exon 9 as compared to the wild type. (C) Representing exon 8, intron 8, and exon 9 in the CLCN7 gene. (D) Showing the cDNA-Sanger sequencing electrogram results of the control. (E) cDNA-Sanger sequencing electrograms of the index, analysis showing 50 bp of intron 8 and exon 9. (F) The green shaded area represents the nucleotides in the intron 8 confirms the presence of intron sequence in index cDNA.](/content/185/2023/35/1/img/10.1016_j.jksus.2022.102377-fig2.png)
(A) Gel image showing the RT-PCR results of the index, father, mother and control samples. Lane (L: 1) showing 100 bp ladder, lane (L:2) index [449 bp], lane (L: 3) father, lane (L: 4) mother, and lane (L: 5) represented the control [399 bp] (healthy individual). The control shows a clear single band size of 399 bp. While the index showed a 449 bp band, suggesting the insertion of 50 bp in the index, both parents showed double bands of 449 bp and 399 bp. (B) Schematic representation of CLCN7 exons and introns. Arrow represents the identified variant's position and retention of the 50 bp sequence of intron 8. As a result, this variant results in large exon 9 as compared to the wild type. (C) Representing exon 8, intron 8, and exon 9 in the CLCN7 gene. (D) Showing the cDNA-Sanger sequencing electrogram results of the control. (E) cDNA-Sanger sequencing electrograms of the index, analysis showing 50 bp of intron 8 and exon 9. (F) The green shaded area represents the nucleotides in the intron 8 confirms the presence of intron sequence in index cDNA.
Retention of the intron may cause disease or regulate gene expression. That happens by introducing additional protein sequences that prohibit the transcript from leaving the nucleus if the retained intron introduces a premature stop codon or might lead to nonsense-mediated mRNA decay (NMD). The trend toward lower CLCN7 levels proposes that intron retention leads to NMD.
RT-PCR of the novel variant abnormality within the CLCN7 gene in osteopetrosis showed significantly increased retention in the homozygous condition of intron 8. The identified splice site variant resulted in the retention of 50 bp of intron 8, thus adding 50 bp extra sequence to exon 9 (Fig. 2 B). Retention of intron 8 results in a premature stop codon in the exon 8 that is 24 bp downstream and is thought to lead to NMD of the mutant transcript or a truncated protein (246 amino acids versus wild-type 805 amino acids).
4.4 Western blot analysis
Western blot was performed to assess the impact of the pathogenicity of the identified mutation at the protein level (Al Mutairi et al., 2020). The fibroblast lysates from the affected individual were subjected to Western blot analysis to detect ClC-7 (Fig. 3A). Chloride-proton exchangers and ClC-type chloride channels exhibit an evolutionarily, distinctive conserved double-barreled structure with two ion conduction pathways created by a single subunit. The functional protein of hetero-dimeric channels comprises a homogenous population of functionally distinct ClC subunits. Western blot analysis showed that the heteromeric form of ClC-7 disappeared in the patient’s fibroblasts. However, the monomeric form corresponding to the standard ClC-7 expression level revealed no significant change in the patient’s fibroblasts. Our findings suggest that the mutation may lead to an anomaly at the binding site, involved in inter-subunit interactions, which impedes the formation of heteromeric ClC channels. This outcome is consistent with the predicted loss-of-function effect of the deep intronic variant, which may be related to an impact of the p.Met250Argfs*6 variant on the protein conformation and stability (Fig. 3B). Thus, this protein expression level analysis further supported the implication of the CLCN7 variant (c.739-18G > A) on our patient’s phenotypic features.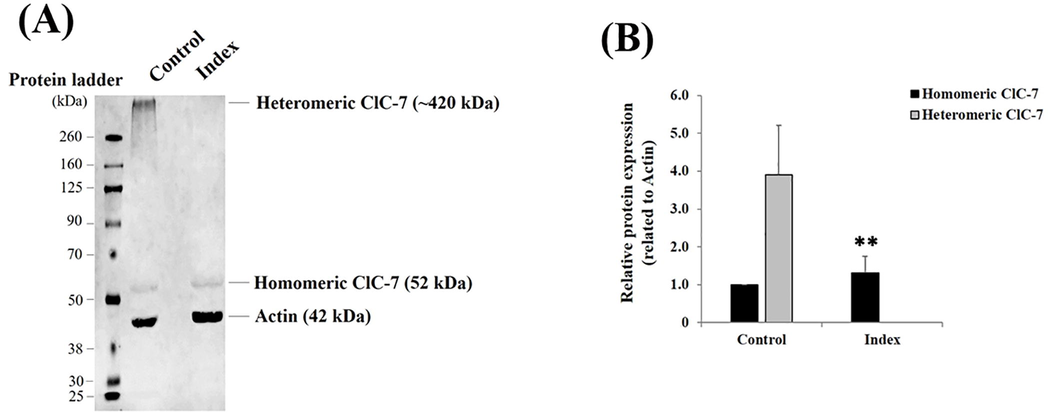
(A) Representative western blot showing the protein expression levels of homomeric and heteromeric ClC-7 detected in fibroblast lysates isolated from healthy individuals (i.e., Control) and patient (i.e., Index). The heteromeric ClC-7 protein expression level fully disappeared in the index compared to the control fibroblasts, whereas the homomeric ClC-7 protein expression level did not change. (B) Bar graph indicating the relative homomeric and heteromeric ClC-7 protein expression levels related to actin, a housekeeping cytoskeletal protein used as a loading control. Data are shown as mean ± standard deviation (SD) (n = 3). **: p = 0.006528 (One-way ANOVA).
5 Discussion
Osteopetrosis is a heterogeneous and rare inherited bone resorption disorder. It represents a cluster of genetic skeletal disorders characterized by increased bone density because of differentiation or functional abnormalities in osteoclasts (Maurizi and Teti, 2019; Bubshait et al., 2020). There seem to be currently at least 11 genes reported to cause osteopetrosis (Zeng et al., 2016). The disease severity and inheritance patterns can be the (benign) form of an autosomal dominant adult considered moderate and maybe exhibit limited symptoms or asymptomatic. The other forms of osteopetrosis include the (malignant) autosomal recessive infantile (Stark and Savarirayan, 2009), the intermediate recessive of a less severe form, and X-linked osteopetrosis (Amirfiroozy et al., 2017).
In this study, we report two unrelated Saudi families showing hallmark features of osteopetrosis. Our genetic testing reports the identification of a novel splice acceptor site variant in the CLCN7 gene (c.739-18G > A). The identified novel variant (c.739-18G > A) interferes with the splicing of the CLCN7 gene [exon 8 and exon 9], that way trimming all the functional domains of the encoded protein. “Loss of function mutations” represents these truncating mutations (Won et al., 2017). Pathogenicity of the identified variant was confirmed using RT-PCR and western blotting analysis.
RT-PCR showed that the variant (c.739-18G > A) altered the splicing pattern of the CLCN7 gene [exon8-exon9], induced the use of a cryptic splice site, and added 50 bp to exon 9, thus leading to a frameshift that caused a premature stop codon (p.Met250Argfs*6) (Fig. 2 C-F). However, the variant is predicted to result in truncated ClC-7 protein, most likely degraded by NMD (Brogna and Wen, 2009).
A genetic change in the DNA sequence happens at the splice site (exon–intron boundary). This alteration may disrupt RNA splicing, causing exon skipping or intron retention and thus altering the protein-coding sequence. Mutations in the region of canonical acceptor and donor sites influence strongly conserved sequences that define exon–intron boundaries. The components of the spliceosome can identify the 3′ splice site (NYAG/G sequence) and 5′ splice site (CAG/GUAAGU sequence) (Agafonov et al., 2016). Furthermore, any variants in these canonical sequences may alter the interaction between proteins involved in the intron removal and pre-mRNA. The most classical pathogenic mutations affect − 1 and − 2 residues at the 3′ acceptor splice site and + 1 and + 2 residues at the 5′ donor splice site. The splice site in the downstream part of an intron [3′ to 5′ direction] is called the splice acceptor splice site, while the splice site in the upstream region is termed the splice donor splice site (5′ to 3′ direction). If the splice site is weak and the presence of mutation reveals the cryptic splice site in a nearby exon or intron, the splicing process can employ this alternate site. This can result in the retention of the intron fragment or the removal of an exon fragment if the cryptic splice site is in the intron or exon, respectively (Anna and Monika, 2018).
Our results illustrated that the heteromeric form of ClC-7 disappeared in the patient’s fibroblasts. However, the monomeric form corresponding to the standard ClC-7 expression level revealed no significant change in the patient’s fibroblasts. This result is suitable for the predicted loss-of-function effect of the variant in deep intronic. In the physiological conditions, characterized by the control phenotype, the chloride channels assemble to homo- or heteromeric dimers, which include sometimes the requirement of accessory β-subunits, for proper function (Jentsch and Pusch, 2018). Furthermore, western blot analyses targeting other CLC members may reveal the molecular composition of this observed high-molecular-weight heteromeric CLC-7 in the normal case (Control).
Several mutations have been reported in osteopetrosis patients from the Saudi population; a five-week-old patient having CAII gene (c.232 + 1G > A) variant, with the initial diagnosis and evaluation was osteopetrosis with secondary renal tubular acidosis (RTA) due to carbonic anhydrase II deficiency syndrome. Also, one of the symptoms that this patient had was the presence of metabolic acidosis (Singh et al., 2021). He suffered poor feeding and thrombocytopenia. Al-Aama et al. (2012) reported a 6-year-old boy diagnosed with osteopetrosis at five months of age, with phenotypes such as poor vision, developmental delay, and hepatosplenomegaly. Molecular diagnosis revealed a homozygous missense variant (c.688A > G; p.Lys230Glu) in the CLCN7 gene (Al-Aama et al., 2012). Similarly, 28 children with autosomal recessive osteopetrosis were reported from two hospitals in Riyadh, Saudi Arabia. Eighteen revealed osteopetrosis with metabolic acidosis, nine showed malignant infantile osteopetrosis, and one patient was mild with delayed onset (Al-Rasheed et al., 1998).
Caetano-Lopes and collages were reported to determine whether the CLCN7 knock-in allele in the mice can produce a mutant protein by studying the RT-PCR, quantified amount of transcripts, and immunofluorescence in the cells. It was observed that the mice lack tooth eruption and die by ∼ 1 month, and at 12 weeks, a slight increase in bone minerals density and differences in cortical bone parameters were observed (Caetano-Lopes et al., 2017). The CLCN7−/− knockout mice exhibited significant retinal and neural deterioration along with diffuse osteopetrosis (Kornak et al., 2001). Rajan et al. (2011) noticed a rapidly progressive loss of the photoreceptor layers and retina's outer nuclear layer in knockout mice. Histological examination of skull and limbs showed normal morphology, and osteoclasts' number or morphologic appearance did not appear to differ from those in wild-type littermates (Rajan et al., 2011). Similarly, Lange et al. (2006) reported that the ClC-7 is significantly reduced in animal models which lack Ostm1, suggesting that the interaction between ClC-7–Ostm1 is necessary for the stability of a protein. In Ostm1-deficient cells and tissues, such as osteoclasts, ClC-7 protein levels reduce below 10 % of normal. Ostm1 mutations were likely to cause osteopetrosis by impairing the acidification environment of the osteoclast depending on the ClC-7 protein level. (Kornak et al., 2001). The finding that animal models, such as ClC-7 deficient mice, display osteopetrosis and neurodegeneration imply more importance of ClC-7–Ostm1 complexes (Lange et al., 2006).
In conclusion, we identified a novel splice site variant in the CLCN7 gene. The findings from the present study increase the spectrum of reported CLCN7 gene mutations and advance our knowledge of osteopetrosis. Additionally, it may aid further studies and future research aiming to explain the heterogeneity of osteopetrosis.
Compliance with ethical standards.
6 Ethical standards
All procedures followed were following the ethical standards and approved by the research committee of King Abdullah International Medical Research Centre in Riyadh, Saudi Arabia.
Acknowledgments
We are grateful to the patients and their families reported in this article for their genuine support. Researchers Supporting Project number (RSP-2021/214), King Saud University, Riyadh, Saudi Arabia.
Funding sources
No funding is involved.
Ethics statement
Written informed consent was obtained from the patient (father).
Author contributions
Klaab Z, and Umair M, drafted the manuscript. Klaab Z, Al Tuwaijri A, and Alfadhel M collected samples of clinical data, analyzed data, and performed experiments. Alfadhel M edited the manuscript. Conception and design of the work: Aldahmash B, Alfadhel M.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Homozygous truncating NEK10 mutation, associated with primary ciliary dyskinesia: a case report. BMC Pulmonary Med.. 2020;20(1):141.
- [CrossRef] [Google Scholar]
- A newly described mutation of the CLCN7 gene causes neuropathic autosomal recessive osteopetrosis in an Arab family. Clin. Dysmorphol.. 2012;21(1):1-7.
- [CrossRef] [Google Scholar]
- Multiple Mitochondrial Dysfunctions Syndrome 4 Due to ISCA2 Gene Defects: A Review. Child Neurol. Open. 2019;6
- [CrossRef] [Google Scholar]
- (a). The Leukodystrophy Spectrum in Saudi Arabia: Epidemiological, Clinical, Radiological, and Genetic Data. Front. Pediatr.. 2021;9
- [CrossRef] [Google Scholar]
- Identification of the TTC26 Splice Variant in a Novel Complex Ciliopathy Syndrome with Biliary, Renal, Neurological, and Skeletal Manifestations. Molecular Syndromology. 2021;12(3):133-140.
- [CrossRef] [Google Scholar]
- A novel mutation in SNX10 gene causes malignant infantile osteopetrosis. Avicenna J. Med. Biotechnol.. 2017;9(4):205-208.
- [Google Scholar]
- Splicing mutations in human genetic disorders: examples, detection, and confirmation. HUMAN GENETICS • REVIEW. 2018;59:253-286.
- [CrossRef] [Google Scholar]
- A. Asiri E. Aloyouni M. Umair Y. Alyafee al Tuwaijri, A., Alhamoudi, K. M., Almuzzaini, B., al Baz, A., Alwadaani, D., Nashabat, M., & Alfadhel, M. Mutated RAP1GDS1 causes a new syndrome of dysmorphic feature, intellectual disability & speech delay Annals of Clinical and Translational Neurology 7 6 2020 956 964 10.1002/ACN3.51059.
- Nonsense-mediated mRNA decay (NMD) mechanisms. Nat. Struct. Mol. Biol.. 2009;16(2):107-113.
- [Google Scholar]
- Malignant Infantile Osteopetrosis: A Case Report. Cureus. 2020;12(1):1-9.
- [CrossRef] [Google Scholar]
- Identification and characterization of a novel CLCN7 variant associated with osteopetrosis. Genes. 2020;11(11):1242.
- [Google Scholar]
- Clcn7F318L/+ as a new mouse model of Albers-Schönberg disease. Bone. 2017;105:253-261.
- [CrossRef] [Google Scholar]
- Genetics, pathogenesis and complications of osteopetrosis. Bone. 2008;42(1):19-29.
- [CrossRef] [Google Scholar]
- The Rare Bone Disease Working Group: report from the 2016 American Society for Bone and Mineral Research Annual Meeting. Bone. 2017;102:80-84.
- [CrossRef] [Google Scholar]
- CLC chloride channels and transporters: Structure, function, physiology, and disease. Physiol. Rev.. 2018;98(3):1493-1590.
- [CrossRef] [Google Scholar]
- Whole exome sequencing identified two novel homozygous missense variants in the same codon of CLCN7 underlying autosomal recessive infantile malignant osteopetrosis in a Pakistani family. Mol. Biol. Rep.. 2018;45(4):565-570.
- [CrossRef] [Google Scholar]
- Loss of the ClC-7 Chloride Channel Leads to Osteopetrosis in Mice and Man. Cell. 2001;104(2):205-215.
- [CrossRef] [Google Scholar]
- A Fatal Case of Infantile Malignant Osteopetrosis Complicated by Pulmonary Arterial Hypertension after Hematopoietic Stem Cell Transplantation. Tohoku J. Exp. Med.. 2014;234(4):309-312.
- [Google Scholar]
- ClC-7 requires Ostm1 as a β-subunit to support bone resorption and lysosomal function. Nature. 2006;440(7081):220-223.
- [Google Scholar]
- Novel CLCN7 mutations cause autosomal dominant osteopetrosis type II and intermediate autosomal recessive osteopetrosis. Mol. Med. Rep.. 2019;19(6):5030-5038.
- [CrossRef] [Google Scholar]
- Therapeutic potential of nanoceria pretreatment in preventing the development of urological chronic pelvic pain syndrome: Immunomodulation via reactive oxygen species scavenging and SerpinB2 downregulation. Bioeng. Transl. Med.. 2022;e10346
- [CrossRef] [Google Scholar]
- Hematopoietic stem cell transplantation corrects osteopetrosis in a child carrying a novel homozygous mutation in the FERMT3 gene. Bone. 2017;97:126-129.
- [CrossRef] [Google Scholar]
- Novel mutations of CLCN7 cause autosomal dominant osteopetrosis type II (ADO-II) and intermediate autosomal recessive osteopetrosis (IARO) in Chinese patients. Osteoporos. Int.. 2016;27(3):1047-1055.
- [CrossRef] [Google Scholar]
- One disease, many genes: Implications for the treatment of osteopetroses. Front. Endocrinol.. 2019;10(FEB):1-9.
- [CrossRef] [Google Scholar]
- An alternative splicing variant in CLCN7-/- mice prevents osteopetrosis but not neural and retinal degeneration. Vet. Pathol.. 2011;48(3):663-675.
- [CrossRef] [Google Scholar]
- Osteopetrosis and renal tubular acidosis: Questions. Pediatric Nephrology (Berlin, Germany). 2021;36(12):4051-4053.
- [CrossRef] [Google Scholar]
- Novel missense loss-of-function mutations of WNT1 in an autosomal recessive Osteogenesis imperfecta patient. Eur. J. Med. Genet.. 2017;60(8):411-415.
- [CrossRef] [Google Scholar]
- Diagnosis and management of osteopetrosis: Consensus guidelines from the osteopetrosis working group. J. Clin. Endocrinol. Metab.. 2017;102(9):3111-3123.
- [CrossRef] [Google Scholar]
- A novel mutation and a known mutation in the CLCN7 gene associated with relatively stable infantile malignant osteopetrosis in a Chinese patient. Gene. 2016;576(1):176-181.
- [CrossRef] [Google Scholar]




