

Translate this page into:
A network pharmacology approach with experimental validation to discover protective mechanism of poly herbal extract on diabetes mellitus
⁎Corresponding author. skmishra.phe@itbhu.ac.in (Sunil Kumar Mishra)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract

Abstract
Objective
Polyherbal extracts (PHE) contain six traditional medicinal plants, and the efficacy of the medicinal plants used in the preparation of this PHE has been confirmed for the treatment of diseases like diabetes mellitus (DM). The aim of this study was to evaluate the efficacy and therapeutic mechanism of PHE through a network pharmacology approach to reveal the protective mechanism of Alpha-Tocospiro A (ATA) present in PHE on DM with experimental validation.
Methods
In this study, Lipinski's rule (Swiss ADME) and drug-likeness score (MolSoft's) web pages were used to confirm the drug-likeness of identified constituents in PHE. Swiss Target Prediction (STP) genes were found for ATA-related genes. The DisGeNet database was used to screen genes associated with DM. String created a network diagram of the interactions between the ATA and DM genes. Top-scoring genes from the string network through CytoNCA plugged into Cytoscape 3.8.2 were selected as hub genes. In addition, the ShinyGO database is used to predict GO and KEGG pathway enrichment analyses.
Results
A total of 675 and 105 therapeutic genes (STP) were associated with all bioactive compounds and ATA in the PHE screen, respectively. Additionally, a maximum of 2,803 DM-related genes (DisGeNet) were observed. Further, in the analysis, 331, 57 potential (intersecting) genes were identified in the correlation between the target genes of all compounds and ATA, respectively, of PHE and the target genes of DM. The identified hub gene “TNF” for both ATA and PHE was found to be precisely strengthened in 49 pathways, along with 14 signaling pathways out of more than 100 enriched KEGG pathways. This study predicted that ATA activates PI3K/Akt and MAPK pathways enriched with TNF by phosphorylating the insulin receptor (IR) β-subunit. The anti-diabetic activity of PHE was found to be good and primarily confirmed by in vitro α-glucosidase enzyme inhibition activity.
Conclusion
The anti-diabetic activity of PHE was found to be effective and was confirmed by the enzyme inhibition activity in the primary study. This study predicted that ATA is a novel drug molecule in PHE that has a targeted mechanism of action and therapeutic effect on DM.
Keywords
Poly herbal extract (PHE)
Alpha Tocospiro A (ATA)
Diabetes mellitus (DM)
KEGG
Molecular simulation
MM/GBSA

1 Introduction
Although immense improvement has been achieved in drug development and pharmaceutical technology over the last century, some polygenic chronic diseases, such as cardiovascular events, brain hemorrhage, cancer, and diabetes mellitus (DM), remain difficult to treat due to their multi-factorial nature (Sagner et al., 2017). DM is rapidly increasing globally, exacerbating its enormous social and economic impact (Seehusen et al., 2019). According to the most recent information available from the International Diabetes Federation (IDF-Diabetes Atlas 2022 Reports), it was believed that a total of 476 million indigenous peoples, hailing from over 5,000 distinct groups and residing in over 90 countries across the globe, would be affected by diabetes. People with diabetes who had HbA1C levels that were greater than or equal to 7 % were 35–40 % more likely to be diagnosed with serious ailments, which include hospitalization, if they acquired COVID-19 (IDF, 2022). Poor glycemic control was a contributing factor to adverse COVID-19 endpoints.
Several mechanisms are involved in the pathogenesis of diabetes, like oxidative stress, insulin resistance, impaired insulin secretion, increased hepatic glucose, and lipid production (Singh et al., 2023). Pancreatic β-cells produce insulin in response to increased blood glucose levels (hyperglycemia), thereby promoting glucose uptake for metabolic activities. The diminished production of insulin, the inability of pancreatic β-cells to meet the required levels, and continuous β-cell failure are believed to be attributed to persistent elevations in plasma glucose concentrations, while strong insulin therapy has demonstrated the ability to induce β-cell responsiveness to insulin, glucagon, and glucose. The stimulation of PI3K/Akt signaling cascades and MAPK pathways enriched with TNF occurs upon the binding of insulin to insulin receptors (by phosphorylating the insulin receptor (IR) β-subunit). Insulin signaling triggers the activation of glycogen synthase kinase 3 (GSK3) in adipose tissues and skeletal muscle cells. This stimulation leads to the inhibition of glycogen production and opens the door for the translocation of vesicles carrying GLUT-4 to the cell membrane, thereby promoting glucose uptake.
A variety of anti-diabetic medications are available on the market today, including GLP-1 receptor agonists, α-glucosidase antagonists, sodium-dependent glucose co-transporter 2 antagonists, dipeptidyle peptidase-4 enzyme blockers, and dopamine-2 agonists, as well as insulin and its analogues (Kerru et al., 2018). The effectiveness of these drugs in the treatment of DM and its complications has yet to be validated, despite the extensive research into their clinical efficacy (Kumar et al., 2016). Some drugs may be able to exert their therapeutic potential across their anticipated drug targets. Whereas, due to their interference with other cellular functions, drugs may also have an influence on the actions of certain interacting cells, resulting in side effects. Because of the increased metabolic correlations between the cellular components, a disease is typically caused by a disruption of the complicated intracellular network rather than being the result of a genetic defect. Thus, medicine's work on intracellular networks for the treatment of disease exerted its therapeutic potential against disease. A network pharmacology study is a new emerging tool that has the potential to make significant contributions to upward drug discovery by providing an opportunity for systematic exploration, which in turn leads to the establishment of disease functionalities and corridors (Fiscon et al., 2018).
Biodiversity products such as traditional herbal formulas, plant extracts, and their bioactive components have recently stimulated the interest of researchers as potential therapeutic targets for diabetes (Xu et al., 2018). These natural products demonstrated their therapeutic action in a manner similar to that of drug candidates by having to interact further with diabetes-related biomolecular markers (Alam et al., 2019). Ayurvedic and traditional medications contain a variety of therapeutic agents that have been shown to be effective in the management of diabetes. Plant-based systems of medicine are predominant in traditional medicine, and individual treatments are frequently based on multiple bioactive constituents that can utilize multiple proteins while influencing numerous pathways, resulting in synergistic impacts on health. A few studies have looked into the use of plant-based medications to help people with DM (Khanal and Patil, 2021). Certain categories of phytoconstituents, such as flavonoids, polyphenols, and alkaloids, have been shown to inhibit the AMP-activated protein kinase (AMPK) (Vazirian et al., 2018), COX-1/-2 (Xu et al., 2020), and dipeptidyl peptidase-4 (Abbas et al., 2019), which are influential in DM. Lignans, ploy-acetylenes, diterpenes, curcuminoids, flavonoids, and amorfruitins (Rigano et al., 2017) have been identified as catalysts of the peroxisome proliferator-activated receptor gamma (PPARγ). Tocopherols play an important role as antioxidants and in maintaining membrane stability in plants. Alpha Tocospiro A (ATA) and alpha Tocospiro B are types of tocopherols that have shown good antioxidant properties (Bano and Deora, 2019). As we all know, phytoconstituents have multi-targeting properties due to their medicinal values (Xu et al., 2018; Zhai et al., 2018), which may be advantageous in the treatment of complex multigenetic diseases such as DM (Poornima et al., 2016) when compared to commercial compounds that are designed to target specific targets. Thus, on the basis of the bioactivities (anti-inflammatory and anti-hyperglycemic) of ATA described in our previous article (Singh et al., 2023), we selected ATA identified in GC–MS profiling of PHE for network pharmacology analysis on the basis of predictive scores, among others.
PHE, derived from the ayurvedic remedy used in this study (Singh et al., 2023), contains ethanolic extracts of six traditional medicinal plants: dried powder of the fruits of Terminalia chebula Retz. (Combretaceae) and Terminalia bellerica Roxb. (Combretaceae); whole herb of Andrographis paniculata Nees. (Acanthaceae); stem of Berberis aristata DC. (Berberidaceae); leaves of Nyctanthes arbostratis L. (Oleaceae); and Premna integrifolia L. (Lamiaceae). A large number of clinical studies have been conducted, and the efficacy of the medicinal plants used in the preparation of this PHE has been confirmed for the treatment of diseases like DM. As a result, the development of diabetes treatments that are both more effective and safer, particularly those derived from medicinal plants, has always been the primary focus of research on a global scale and continues to be so at the present time. Synergy between multiple compounds, multiple pathways, and multiple targets is typical of PHE because these combinations are typically what make up PHE. But this complexity also means that the efficacy of individual drugs is uncertain and the mechanism of action is not well understood (Chen et al., 2019). With systemic, collaborative, and meta-analysis modes, network pharmacology is a technique that can be used to explore the association of networking elements like genes, compounds, proteins, and target diseases. The network pharmacology study for natural products is one of the most important sources of multiple targets for a single drug. A model switch from one target to many targets and a concept switch from one drug to many are some of the important aspects of this study. When it comes to uncovering the delicate mechanisms of drug-target relations, network pharmacology has opened a new avenue (Oh et al., 2021). Since the synergistic effects of compounds with biological activity are often unclear, network pharmacology is a useful tool for elucidating these relationships (Luo et al., 2020).
Since our previous research showed that PHE contained multiple bioactive ingredients and had good antioxidant as well as anti-inflammatory activity, we hypothesized that an in silico analysis based on a network pharmacology approach for the mechanisms of action of the bioactive compounds in PHE would shed light on how to best combat DM, as verified by experimental work. Therefore, the present study explained how to construct a hub gene via the network pharmacology approach of ATA in PHE, which attenuates diabetes via multiple pathways focused on activation of PI3K/Akt and MAPK, key signaling pathways in the present network pharmacology study, and provides important evidence for the therapeutic value of ATA in PHE against DM by suppressing free radical oxidative stress, inflammation, and the α-glucosidase enzyme.
2 Materials and methods
2.1 Preparation of polyherbal extract (PHE)
In the present study, the method of preparation of PHE was utilized from our previous report (Singh et al., 2023). The dried powders of biological sources of all selected medicinal plants, 50 g each, were defatted with 400 mL of hexane separately, with occasional shaking for 72 h. After defatting, the maceration method of extraction was carried out for 72 h with the above completely dried powdered material in ethanol (200 mL) as a solvent with occasional shaking. Following filtering, rota-vapor was used to evaporate the residue to a dryness of less than 50 °C, and the crude extracts obtained from these medicinal plant materials were used to prepare PHE by mixing in equal amounts. Properly mixed, prepared, and close-packaged PHE was used for characterization in the next steps. Yield of each extract, a list of the bioactive phytoconstituents of PHE identified by gas chromatography-mass spectrometry (GC–MS) analysis, and a more significant level of antioxidants as well as in-vitro anti-inflammatory activity of PHE were also reported in our previous study, which stated that PHE has a synergistic effect (Singh et al., 2023).
2.2 Network pharmacology-based study
2.2.1 Compounds database construction and drug-likeness filtering
In the previous GC–MS analysis of PHE, the identified bioactive compounds present in PHE were used for this study. Prediction of ADMET, drug likeness score (DLS), and side effects of individual phytoconstituents was performed using admetSAR2.0 (https://lmmd.ecust.edu.cn/admetsar2), MolSoft (https://molsoft.com/mprop/), and ADVERPred (https://www.way2drug.com/adverpred/), which were used to calculate ADMET characteristics, drug likeness score, and probable side effects, respectively. To obtain the SMILES of phytoconstituents, the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) was used (Roman et al., 2019). ATA of PHE was selected for further study on the basis of the above screening results as well as reported biological evaluations in a variety of peer-reviewed publications.
2.2.2 Target genes related to selected compounds and DM
There were some databases at web search engines such as Swiss Target Prediction (STP) with credentials (https://www.swisstargetprediction.ch/) utilizing the option “Homo sapience” to predict genes involved in the preferred compounds based on SMILES. Utilizing another database, DisGeNET (https://www.disgenet.org/), DM genes were retrieved by searching websites. The Venny 2.1 tool, online with credentials available (https://bioinfogp.cnb.csic.es/tools/venny/), was used to pick the genes encoding for compounds. The tool was also used for the genes of DM encoding, recognition, and visualization (Agrawal et al., 2023).
2.2.3 Network construction of interconnection between active ingredients and overlapping genes
The number of nodes and edges was determined by calculating the correlations among both compounds and the related genes based on STP results. In this study, the network visualization of target genes was created by String (https://string-db.org/). Nodes in the network are representations of the compounds and genes, while associations among both are represented by edges (Kumar et al., 2022a).The bioactive compounds and hub genes of PHE as well as individual compounds against diabetes were defined by implementing the “degree value (DC), closeness (CC), and betweenness (BC) without weight” of compounds and genes acquired by establishing the topological features of networks with CytoNCA plugged into Cytoscape 3.8.2 (Kumar et al., 2022b). The total number of edges for compounds as well as genes at the network scale is described mostly by the degree value obtained. The higher the degree values of compounds or genes, the greater the therapeutic benefit (Lee et al., 2018) of the ATA of PHE on diabetes was noted.
2.2.4 Pathways and interactive analysis of hub genes
The “Homo sapiens” mode of ShinyGO v0.741: “Gene Ontology Enrichment Analysis + More” database, with a search option for genes on the web (https://bioinformatics.sdstate.edu/go/), was used to conduct a KEGG pathway enrichment analysis of intersecting genes recognized from the venn diagram (Filimonovet al., 2014). The results of the KEGG enrichment pathway analysis were used to infer the probable molecular mechanisms of PHE on DM (Filimonov et al., 2014). The lollipop chart of pathways was plotted using the ShinyGO v0.741 “Gene Ontology Enrichment Analysis + More” database to represent the potential molecular mechanism pathways of the ATA of PHE to overcome DM.
2.3 Prediction of biological spectrum
By using the Prediction of Activity Spectra (PASS) database for compounds, it was possible to predict the anticipated biological spectrum of phytoconstituents. The probable functions were denoted by the letters “probable activity” (Pa) and “probable inactivity” (Pi). The activities associated with Pa > Pi were taken into consideration in the current analysis of biological spectrum interpretation (Gligorić et al., 2020).
2.4 Molecular docking study
2.4.1 The binding affinity energy analysis of the highest scored ingredient on a hub gene
Autodock 4.2.6 software (https://autodock.scripps.edu/) was utilized to estimate the molecular docking energy of the primary ingredient on hub genes (Balogun et al., 2022).
2.4.2 Molecular dynamics simulation analysis
To simulate classical molecular dynamics (MD), the autodocked complexes were subjected to MD simulation using Desmond software, which is part of the Schrödinger suite of programs (Khanal and Patil, 2021). The clear and direct solvent systems were included in the MD simulation study when they were created. An orthorhombic box (10x10x10 buffer), TIP4P (transferable intermolecular potential-4 points), and 0.15 M salt were used to construct the model, which was then improved to better represent physiological conditions. A total of 300 K and 1 atm of pressure were utilized to neutralize the entire simulation model, which was then neutralized by incorporating counter-sodium and chloride ions. In the end, the two different autodock complexes were designed to simulate for a 100 ns time frame under default parameters using the OPLS-2005 force field. The results were compared with the experimental results. At 10 ps intervals, 10,000 frames were taken (Khanal and Patil, 2021), and a snapshot was taken through each 100 ns MD trajectory. It was then used to look again at the trajectories for the root-mean-square deviation (RMSD), root-mean square fluctuation (RMSF), ligand torsion profiles, and protein–ligand interaction profiling.
2.4.3 Molecular mechanics/generalized born surface area calculation
In this study, the theory of molecular mechanics has been expanded. It was decided to use the Born surface area (MM/GBSA) measurement, which is a highly developed quantum mechanics measurement. With the help of this technique, false-positive results were removed from molecular docking analysis, allowing us to calculate the comparative binding free energy of both the AKT1-ATA complex and the TNF-ATA complex, respectively.
2.5 Experimental validation
2.5.1 In-vitro α-glucosidase inhibition assay
The method described by Bhatia et al. (2019) was utilized in this study to determine whether or not PHE possessed α-glucosidase inhibitory activity. PHE (50 µL) of varying concentrations (from 20 to 140 µg/mL) was kept at 37 °C for 20 min for incubation with the α-glucosidase (maltase) ex. Saccharomyces enzyme solution (1 U/mL) of volume 10 µL, and then a 125 µL solution of 0.1 M phosphate buffer was added to the mixture (pH 6.8). After waiting for 20 min, the reaction was kicked off by adding 20 µL of 1 M pNPG (substrate), and now the solution was allowed to incubate for another 30 min. The reaction was stopped when 50 µL of 0.1 N Na2CO3 was mixed, followed by the measurement of the absorbance of the final solution with a UV–visible spectrophotometer at 405 nm (Shimadzu 1800). At concentrations ranging from 20 to 140 µg/mL, acarbose was employed as a standard drug (positive control) throughout the study. The following is how the enzyme activity was calculated:
Each unit of the enzyme is the same as the quantity of α-glucosidase that is required to produce a quantity of one micromole per minute of the formula p-nitrophenol, which is synthesized from the substrate p-nitrophenyl-α-D-glucopyranoside. A regression equation was obtained by plotting concentrations with a range of 20–140 µg/mL (on the x-axis) and percent inhibition (on the y-axis) for PHE. This allowed for the calculation of IC50, which is the concentration needed to inhibit 50 % of the enzymatic activity.
3 Results
3.1 Network pharmacology-based analysis
3.1.1 Potential active ingredients from poly herbal extract
A maximum percent yield (34.03 for Terminalia chebula) and a minimum percent yield (2.48 for Berberis aristata), among others, were reported in our previous work. The mass peak data of all compounds analyzed in the previous GC–MS study are provided in Supplementary Figs. S1.1 and S1.22 intended with the mass peak data of ATA. The Swiss ADMET database predicted results in the form of Lipinski's rule for all these compounds, confirming that these compounds possess drug-like properties. As we used 3 online web servers (STP, SEA, and Targetnet) to perform the target identification of bioactive molecules, a deficit in the targets of some compounds and interactions between all the screened compounds of PHE were found with the online servers SEA and Targetnet. Thus, the STP database was the most suitable server among the others and was used to select targets from all the screened compounds that interacted with the genes in the present study. These interacted compounds were considered to be the therapeutic components among them. Consequently, among all the compounds of PHE, only compound ATA resulted as the most suitable for further network pharmacology study because it had a comparatively higher (0.22) drug-like score (DLS) from the MolSoft server (Supplementary Fig. S2), no side effects predicted from the ADVERPred database (Supplementary Table S1), valuable predicted ADMET properties (Fig. 1A), and also cleared all crucial parameters from the Admet SAR 2.0 (Table 1). Therefore, ATA is found to be a novel drug that is being utilized for the first time in a network pharmacology study against DM.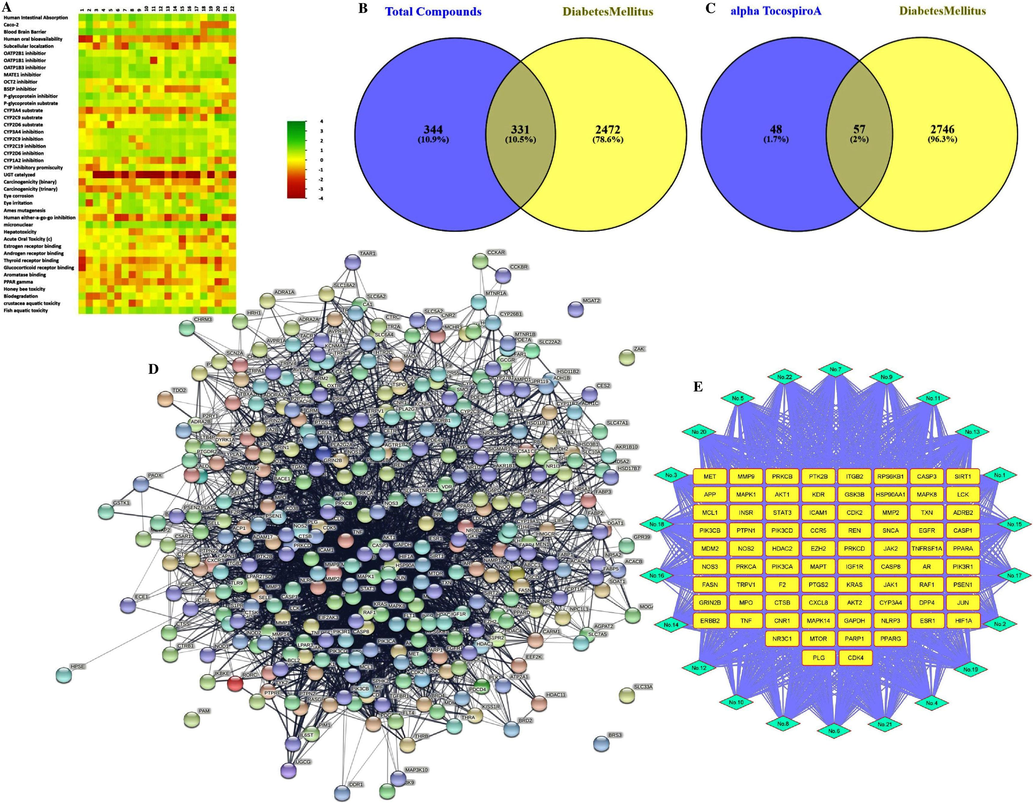
Potential active ingredients of PHE, core targets identification and network construction for the treatment of DM. A) ADMET score of all the compounds to have drug like property, B) Potential 331 core targets identification between 675 STP genes related to all compounds and 2803 DisGenet genes related to DM, C) Potential 57 core targets identification between 105 STP genes related to ATA compound and 2803 DisGenet genes related to DM, D) PPI network of PHE, and E) Compound target network of PHE with top selected genes, light blue diamond shapes represent core compounds and yellow squares represent important potential targets.
S.N.
Name of Compound
CID
MF
MW (g/mol)
NHBA
NHBD
MolLogP
BBB
DLS
1.
Phenol, 2,4-bis(1,1-dimethylethyl)-
7311
C14H22O
206.32
1
1
5.22
4.86
−1.24
2.
2-Buten-1-ol, 2-ethyl-4-(2,2,3-trimethyl-3-cyclopenten-1-yl
119,898
C14H24O
208.34
1
1
4.37
4.36
−0.40
3.
1,2,5,6-Tetrahydrobenzonitrile
66,013
C7H9N
107.15
1
0
2.05
3.62
−1.73
4.
Neophytadiene
10,446
C20H38
278.52
0
0
8.48
1.34
−1.22
5.
4-Piperidinamine, 2,2,6,6-tetramethyl-
37,524
C9H20N2
156.27
2
2
0.53
3.75
−1.10
6.
3,7,11,15-Tetramethyl-2-hexadecen-1-ol
5,366,244
C20H40O
296.53
1
1
7.72
4.31
−0.87
7.
Hexadecanoic acid, methyl ester
8181
C17H34O2
270.5
2
0
7.36
4.53
−1.31
8.
Benzenepropanoic acid, 3,5-bis(1,1-dimethylethyl)-4-hydroxy-, methyl ester
62,603
C18H28O3
292.4
3
1
4.77
4.60
−1.21
9.
1,2-Benzenedicarboxylic acid, dibutyl ester
3026
C16H22O4
278.34
4
0
4.39
4.18
−0.38
10.
13-Hexyloxacyclotridec-10-en-2-one
566,650
C18H32O2
280.4
2
0
6.52
4.52
−0.95
11.
9,12-Octadecadienoic acid, methyl ester
5,284,421
C19H34O2
294.5
2
0
7.32
4.51
−1.04
12.
6-Octadecenoic acid, methyl ester, (Z)-
5,362,717
C19H36O2
296.5
2
0
7.82
4.51
−1.04
13.
Phytol
5,280,435
C20H40O
296.5
1
1
7.72
4.31
−0.87
14.
Methyl stearate
8201
C19H38O2
298.5
2
0
8.37
4.51
−1.31
15.
9-Octadecenoic acid, 12-hydroxy-, methyl ester, [R-(Z)]-
5,354,133
C19H36O3
312.5
3
1
6.39
4.13
−0.68
16.
Undecanoic acid, 5-chloro-, chloromethyl ester
543,301
C12H22Cl2O2
269.20
2
0
4.83
4.47
−0.93
17.
cis-9-Hexadecenal
5,364,643
C16H30O
238.41
1
0
6.54
4.41
−1.25
18.
15-Hydroxypentadecanoic acid
78,360
C15H30O3
258.40
2
2
4.40
3.56
−0.13
19.
Dioctyl phthalate
8346
C24H38O4
390.6
4
0
8.47
4.29
−0.43
20.
1-Glyceryl ricinoleate
5,319,880
C21H40O5
372.5
5
3
5.18
2.23
−0.07
21.
Alpha Tocospiro A
21,674,156
C29H50O4
462.7
4
1
7.96
3.40
0.22
22.
Isopropyl linoleate
5,352,860
C21H38O2
322.5
2
0
7.99
4.47
−0.32
3.1.2 Target genes associated with the compounds and DM
It was observed that a total of 675 genes were associated with all the bioactive compounds in the PHE screen, and 105 genes were associated with the bioactive ingredient ATA by STP databases. Additionally, a maximum of 2,803 DM-related genes were observed by searching DisGeNet databases. By analyzing Venn diagram (Fig. 1B), 331 intersecting genes were identified in the correlation between genes of all compounds of PHE and DM-related genes. Further, it was also identified that 57 intersecting genes (Fig. 1C) were observed between ATA and DM-related genes.
3.1.3 Hub genes and essential active ingredients of Poly herbal extract against DM
The above intersecting genes were studied for network construction by the String database with 0.400 confidence as an analysis parameter and visualized. All selected compounds of PHE manifested 331 nodes and 4272 edges in the network pharmacology analysis. This data revealed the presence of 25.8 average node degrees, a 0.45 average local clustering coefficient, a 1658 expected number of edges, and a p-value of 1.0e-16 for PPI enrichment in the network. It was noticed that some valuable gene such as PAM, BRS3, SLC33A1, ZAK, and MGAT2, have an unconstructed role in the network directly, and such genes were excluded from further study. Based on network pharmacology analysis in reference to the nodes, the edges, and the degree centralities (DC) values of genes from CytoNCA plugged into Cytoscape, a total of 331 genes for all identified PHE compounds may be found to be effective against diabetes with synergistic effects.
Consequently, it was determined that the genes AKT1, TNF, GAPDH, EGFR, STAT3, JUN, CASP3, PPARG, HSP90AA1, ESR1, HIF1A, CXCL8, and MMP9 manifested a higher degree value than others in the network (Supplementary Table S2). These genes served as the hub genes for protection from diabetes. The network generated by string recommended that the clinical manifestations of PHE on diabetes be closely intertwined with the 331 genes observed in the PPI network of PHE (Fig. 1D) as well as all compounds collectively observed in the compound target network (Fig. 1E). Table 2 showed the preferable DC value of ATA from the network pharmacology study for the treatment of DM, along with all the screened compounds of PHE by cytoscape in this study. The ingredients compounds in PHE like 1-Glyceryl ricinoleate, 15-Hydroxypentadecanoic acid, and Methyl stearate also have equal or higher degree values (32, 32, and 37, respectively) compared to ATA (32) as shown in Table 2. Due to the lack of some crucial parameters of drug likeness, these compounds (Table 1) were excluded from further study.
S.N.
Name of Compound
Value
1.
Phenol, 2,4-bis(1,1-dimethylethyl)-
26
2.
2-Buten-1-ol, 2-ethyl-4-(2,2,3-trimethyl-3-cyclopenten-1-yl
11
3.
1,2,5,6-Tetrahydrobenzonitrile
8
4.
Neophytadiene
15
5.
4-Piperidinamine, 2,2,6,6-tetramethyl-
12
6.
3,7,11,15-Tetramethyl-2-hexadecen-1-ol
9
7.
Hexadecanoic acid, methyl ester
22
8.
Benzenepropanoic acid, 3,5-bis(1,1-dimethylethyl)-4-hydroxy-, methyl ester
17
9.
1,2-Benzenedicarboxylic acid, dibutyl ester
14
10.
13-Hexyloxacyclotridec-10-en-2-one
17
11.
9,12-Octadecadienoic acid, methyl ester
23
12.
6-Octadecenoic acid, methyl ester, (Z)-
27
13.
Phytol
9
14.
Methyl stearate
37
15.
9-Octadecenoic acid, 12-hydroxy-, methyl ester, [R-(Z)]-
26
16.
Undecanoic acid, 5-chloro-, chloromethyl ester
28
17.
cis-9-Hexadecenal
26
18.
15-Hydroxypentadecanoic acid
32
19.
Dioctyl phthalate
20
20.
1-Glyceryl ricinoleate
32
21.
Alpha Tocospiro A
32
22.
Isopropyl linoleate
31
3.1.4 Network construction, hub gene analysis and functional annotation of alpha Tocospiro A against DM
With the help of the Venny 2.1 tool online, the genes encoding for ATA and the genes of DM were picked, recognized, and visualized. 57 intersected genes were found for PPI network construction (Fig. 2A). The compound target disease network was visualized using data from the STRING database (Fig. 2B). It was a net gain of 57 nodes and 175 edges, with 6.14 an average node degree, 0.489 an average local clustering coefficient, 55 an expected number of edges, and a p-value of <1.0e−16 PPI enrichment from the network pharmacology analysis of ATA and DM intersected genes. In this network pharmacology analysis, some intersecting genes, such as MAP3K9, SMO, CHRM3, P2RX3, BRS3, ATP2A1, and SLC33A1, were excluded from the study due to a lack of direct network construction. Based on the higher DC values of genes than others in the network (Supplementary Table S3) and network pharmacology analysis data, genes such as TNF, CASP3, SIRT1, HSP90AA1, MAPK1, CASP8, MDM2, MAPK14, MCL1, MAPK8, CASP1, IGF1R, and CDK2 were considered hub genes for ATA against DM (Fig. 2C). With the selection of hub genes, the further activities of these genes were analyzed by utilizing the Gene Ontology (GO) study and the gene enrichment analysis with KEGG pathways by using the ShinyGO v0.741 database. Molecular function (MF; Fig. 2D), biological process (BP; Fig. 2E), as well as cellular component (CC; Fig. 2F), the three valuable classes with detectable properties for target hub genes, were collected from the GO study. The GO detected properties for target hub genes such as apoptotic signaling pathways, catalytic activity, protein domain specific binding, cellular response to environmental stimulus as well as chemical stress, regulation of apoptotic process, death-inducing signaling complex, mitochondrion, nucleoplasm, and nuclear lumen from all three valuable classes show higher values than other important predicted values in reference to FDR and number of genes. It was determined that the selected hub gene TNF, which manifested with the highest degree value, i.e., 32, served as the top hub gene of ATA for protection from DM, and it was also the hub gene of PHE with the 2nd highest DC value (162) as observed in the above study. Thus, the top two genes, AKT1 (167) and TNF (162), were selected for further studies.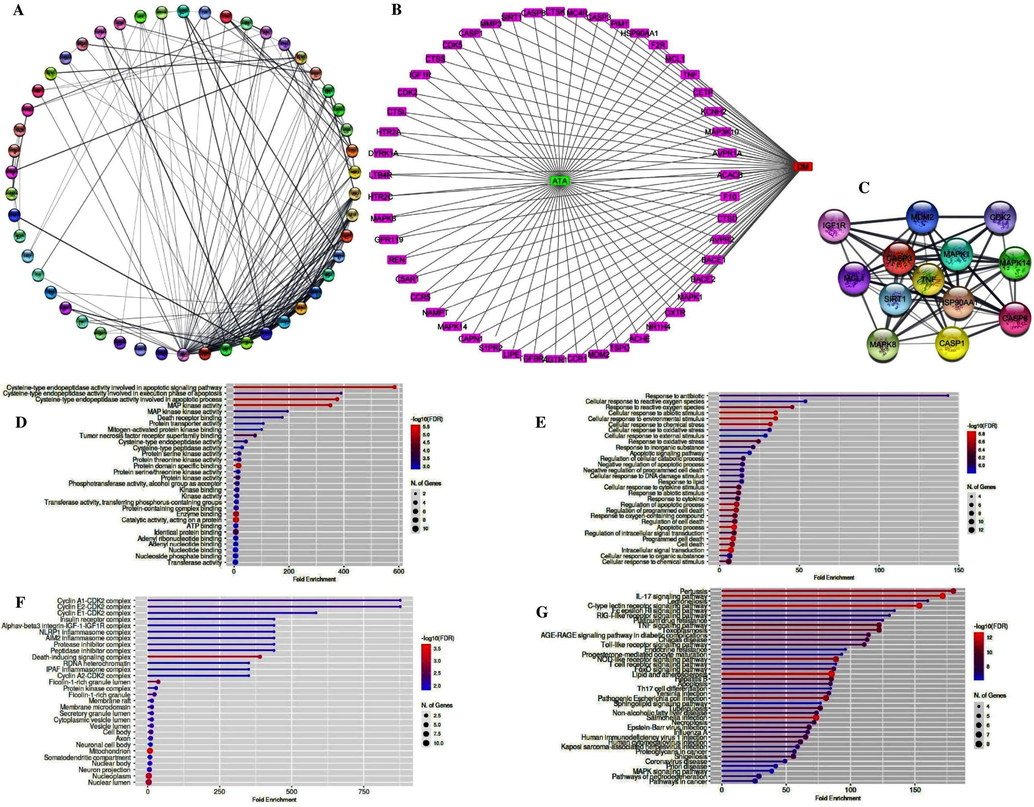
Network construction, hub gene analysis, functional annotation and potential molecular pathways of ATA for the treatment of DM. A) PPI network of ATA, B) Compound target disease network of ATA, green square represents important potential core compound-ATA, pink rectangles represent important potential targets and red square represents DM. C) Top 13 selected hub genes network of ATA, D) Molecular functions, E) Biological processes, F) Cellular components, G) Top most KEGG pathway enrichment in study of ATA.
3.1.5 Potential molecular pathways of alpha Tocospiro A against DM
Following the KEGG pathway enrichment analysis, the top intertwining genes of ATA with network topologies genes of DM were significantly enriched in 100 pathways with P values, i.e., p < 0.05, with 29 signaling pathways being the most abundant; the top pathways were selected on the basis of FDR enrichment value and shown in Fig. 2G. Supplementary Table S4 provides extensive information on pathways that contribute to the development and progression of diabetes. Aside from that, in this study, the hub gene “TNF” was found to be precisely strengthened in 49 pathways, along with 14 signaling pathways. TNF plays a major role in all 49 pathways studied by metabolic pathways, inferring that the TNF signaling pathway (Fig. 3A) might be the hub signaling pathway, along with the JNK/MAPK/ERK and IRS/PI3K/AKT-GLUT4 signaling pathways, for PHE against diabetes, highlighted in Supplementary Table S4. Thus, the study predicted that, ATA stimulates PI3K/AKT and MAPK pathways enriched with TNF by phosphorylating the insulin receptor (IR) β-subunit, which results in glucose homeostasis in different cascades. Furthermore, a crucial and noticeable scenario was also predicted here: the hub genes of ATA were also involved in the present COVID-19 pandemic management, with enrichment of 5 genes (MAPK14, MAPK1, MAPK8, TNF, and CASP1) through the Corona virus disease pathway (hsa05171) in this study.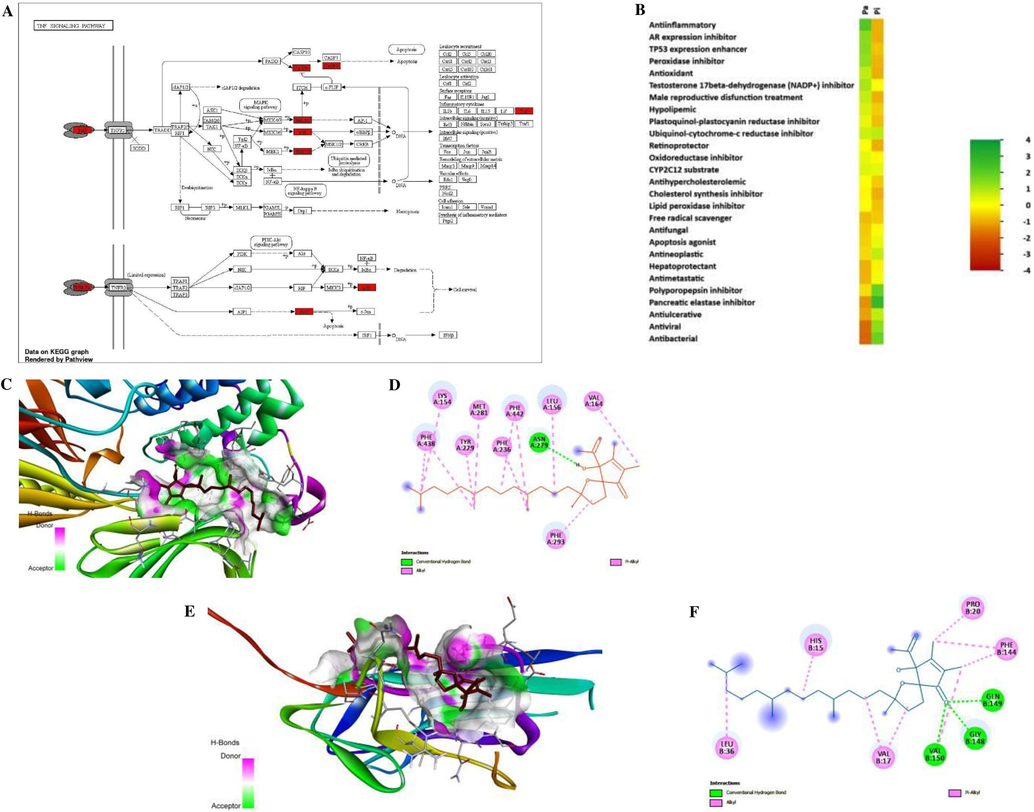
KEGG pathway enrichment, heat map of PASS activity and molecular docking diagram of the ATA with key targets. A) TNF as hub signaling pathway from KEGG pathway enrichment, B) Heat map of top probable bioactivities in the PASS study, C) 3D interaction diagram with AKT1, D) 2D interaction diagram with AKT1, E) 3D interaction diagram with TNF, F) 2D interaction diagram with TNF.
3.2 Activity spectra for substances (PASS)
PASS online prediction revealed different crucial data in Supplementary Table S5, which showed the anticipated activities for the bioactive constituent ATA. Bioactivities like anti-inflammatory, antioxidant, hypolipemic, retinol protector, antihypercholesterolemic, cholesterol synthesis inhibitor, antineoplastic, hepatoprotectant, antiviral, etc. are primarily detected for ATA in this study (Fig. 3B). Anti-inflammatory activity had the greatest predicted effectiveness. As demonstrated by the findings, the probable activity (Pa) values were quite close to 1 and the probable inactivity (Pi) values were extremely close to 0, indicating that the selected bioactive constituent ATA is highly likely to be engaging in these activities.
3.3 Molecular docking analysis
3.3.1 Validation of top selected hub genes protein
Receptor proteins were validated using a Ramachandran plot evaluation. When the plot for AKT1, the top hub gene, was generated, it showed that 92.8 percent of amino acids remained present in the preferred zone (Fig. S3). The plot for TNF-α was generated, and it showed that 91.9 percent of amino acids persisted in the preferred zone (Fig. S4). The ProSA web was also utilized to evaluate the quality of the conformations of the selected proteins (AKT1 and TNF-α). The Z-score map of PDB proteins is scientifically verified by NMR spectroscopy (deep blue) as well as X-ray diffraction (pale blue). The Z-score of the chosen proteins AKT1 (−7.28) and TNF-α (−4.3) is shown in Figs. S5 and S6, respectively.
3.3.2 Affinity binding score of a key bioactive ingredient alpha TocospiroA on hub genes protein
AutoDock 4.0 was utilized to estimate affinity-binding energy for hub genes. The most significant compound, ATA, was docked with hub genes (AKT1; PDB ID 6hhg; and TNF-α; PDB ID 6op0). With the grid point spacing of 0.575 Angstroms, 126-X, 126-Y, 126-Z, and 13.601, −11.818, and −15.800 as Central Grid Point coordinates, the binding energy score for chain A of 6hhg against ATA was −6.99 kcal/mol, and through 0.486 Angstroms grid point spacing, 126-X, 126-Y, 126-Z, and −0.559, −0.686, and 27.615 as Central Grid Point coordinates, the binding energy score for chain B of 6op0 against ATA was −6.05 kcal/mol. As shown in Supplementary Table S6, the binding energy scores of chain A of 6op0 against ATA (−4.80 kcal/mol) and chain C of 6op0 against ATA (−5.59 kcal/mol), as well as the binding site residues on hub genes, were calculated. Discovery Studio was utilized for structure visualization in 3D and 2D. The most important residues on the AKT1 and ATA complex with hydrophobic interaction are LYS154, VAL164, MET281, TYR229, PHE236, PHE293, PHE438, LEU156, PHE442, and also ASN279 with hydrogen bond interaction. The relevant amino acids for chain B of the TNF-α and ATA complex are shown in Supplementary Table S7. The most important amino acid residues for chain B of the TNF-α and ATA complex with hydrophobic interactions are VAL17, PRO20, VAL150, LEU36, HIS15, and PHE144 along with hydrogen bond interactions GLY148, GLN149, and VAL150. 3D and 2D structures for the AKT1 complex with ATA were illustrated in Fig. 3C and D, and the structures for the TNF-α complex with ATA were illustrated in Fig. 3E and F, along with Supplementary Figs. S7 and S8.
3.3.3 Molecular dynamics simulation analysis
To further investigate the compounds' conformational stability, intermolecular interaction profiles, and binding site occupancy, the molecularly docked complexes were subjected to an explicit solvent molecular dynamics (MD) simulation for duration of 100 ns. The formation of intermolecular contacts between the ligand (ATA) and amino acid residues in the active site of hub gene protein (AKT1) resulted in the compound forming one hydrogen bond interaction with ASN279 (Fig. 4A), and the formation of intermolecular contacts between the ligand (ATA) and amino acid residues in the active site of hub gene protein (TNF), shown in Fig. 4B, revealed that the compound forms three hydrogen bond interactions with GLY148, GLN149, and VAL150. Furthermore, in addition to the covalent contact interactions, numerous different non-covalent contact interactions (Fig. 4A–D), including hydrophobic, ionic, and salt bridge interactions, were also indicated by this study. The RMSD values for the protein (Cα) and protein fit ligand (Fig. 4E and F) were recorded from each 100 ns simulation trajectory using the PHE ligand ATA and extrapolated out of each 100 ns simulation trajectory. After complexes with ATA, the Cα-atoms in AKT1 (Fig. 4E) demonstrated significant variance (<3), and the RMSD remained relatively stable within an acceptable range of 2.6–2.9Ǻ from 90 to 99 ns, which is acceptable for small globular proteins during the 100-ns simulation period. Moreover, the Cα atoms of TNF-α (Fig. 4F) docked with ATA showed relatively stable RMSD values ranging from 1.4 Å to 2.3 Å from 7 to 30 ns, followed by slight elevation and a state of equilibrium with a calculated mean of 2.35 ns at the end of the simulation.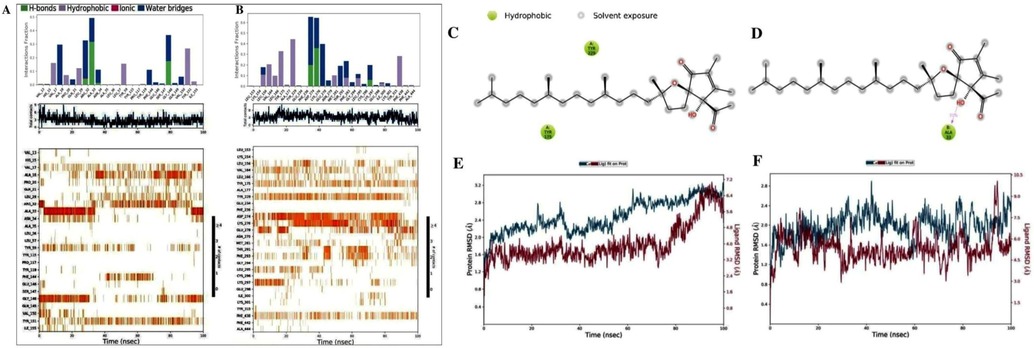
Molecular dynamics simulation analysis diagrams. The stacked bar charts for protein–ligand interaction map. A) AKT1-ATA complex and B) TNF-ATA complex, 2D molecular contacts profiling for the hydrophobic (green) poses. C) AKT1 and D) TNF, and Calculated RMSD values for protein fit ligand ATA (red curves) and alpha carbon (Cα) atoms (blue curves). E) AKT1 and F) TNF.
During the 100 ns MD simulation interval, the PHE ligand exhibits a structural conformational deviation of less than 5 Ǻ which is acceptable. Most notably, the compound ATA (4.9Ǻ and 5.0Ǻ) exhibited the least amount of unacceptable deviation as well as equilibrium throughout the entire modeling process. As a result, the bioactive compound ATA derived from PHE was identified as having significant structural stability in the specific pockets of AKT1 and TNF-α respectively. The local changes and flexibility of AKT1, TNF-α and the ligand ATA in their respective complexes were evaluated for root mean square fluctuations (RMSF) analysis throughout the 100 ns simulation trajectories (Fig. 5A and B). The N (amino) and C (carboxy) terminals of proteins fluctuate more than in other regions. The alpha helices and beta strands tend to be more rigid, thereby fluctuating less than the loop region. The fluctuations were a bit high at 60 ns, with a range between 92 and 95 ns for the simulation times for the docked ligand ATA of TNF-α. Based on the resulting output, the amino acid residues of AKT1 and TNF-α in their respective docked complexes with the ligand ATA demonstrated similar fluctuations (RMSF: about 2.0–5.5 Å) during the simulation study of 100 ns in a region such as indicated for molecular interaction with the ligand, the loop region, and the c-terminal for AKT1 as well as TNF-α. The calculations show that the docked ligand is more stable in the active sites of AKT1 and TNF-α during the 100 ns MD simulation.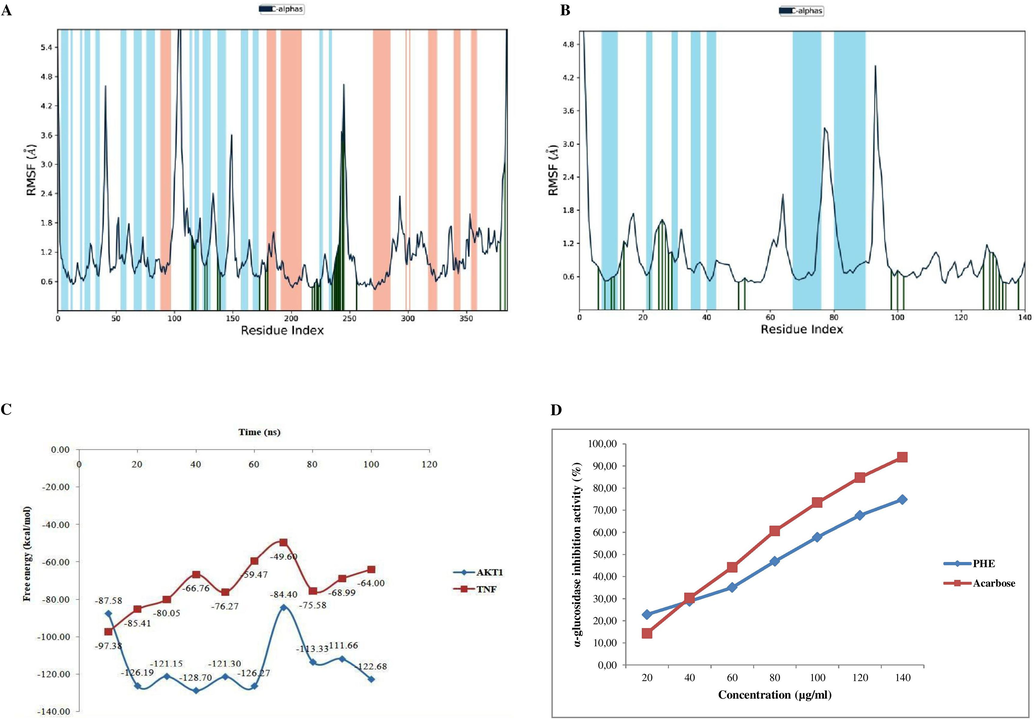
Line representation of the evolution of Root mean square fluctuation (RMSF) of top selected hub genes with Cα – ATA complexes A) AKT1 and B) TNF, C)Binding free energy analysis on the average structure from each cluster of hub gene protein complexes AKT1-ATA (blue), and TNF-ATA (red) calculated using Prime MM/GBSA, D) α-glucosidase inhibitory activity of PHE. Data are expressed as mean ± SD of duplicate determinations.
3.3.4 Molecular mechanics/generalized Born surface area analysis
In the end, docked ligand ATA complexes were used to do relative free binding energy calculations with the MM/GBSA method to figure out the gross receptor-ligand interaction energy between them (Supplementary Table S7). AKT1-ATA complexes docked in the docking chamber demonstrated significant binding free energy (−128.70 kcal/mol) as well as TNF-ATA (−97.38 kcal/mol) values, which were computed, respectively, for the highest and lowest binding energy values. The MM/GBSA raw data of the overall study were extrapolated, and a comparative graph was plotted (Fig. 5C).
3.4 Experimental validation
3.4.1 In-vitro anti-diabetic activity
As shown in Fig. 5D, acarbose, as a reference drug, had a higher inhibitory activity for α-glucosidase compared to PHE. In the present study, PHE showed its maximal α-glucosidase inhibitory activity at 74.84 ± 0.01 % at 140 µg/ml with an IC50 of 85.09 µg/ml and acarbose at 93.94 % at 140 µg/ml with an IC50 of 69.13 µg/ml, as shown in Supplementary Table S8.
4 Discussion
As previously stated, the classical drug research process is predicated mostly on the “single drug-protein-disease theory,” which has been proven to be factually inaccurate more than once, as a single drug moiety can engage different proteins as well as modify multiple pathways (Lee et al., 2018). A higher number of single-target drugs (Filimonov et al., 2014) may be expected to have great therapeutic effects, which are frequently associated with more than just a variety of side effects. To reduce this risk, it is possible to treat the disease pipeline by considering all possible proteins in order to build up an additive or synergistic impact. Thus, a more holistic treatment method can be successfully accomplished for these complications via alternative traditional systems of medicine. It includes the use of medicinal herbs containing different bioactive constituents to target proteins involved in modifying disease-associated mechanisms in a variety of different genetic scenarios and disease etiologies (Khanal and Patil, 2021). Complete management of DM by PHE may be a good approach for treatment with herbal medicine.
ATA was found to be a novel compound in PHE (Singh et al., 2023). ATA and alpha-Tocospiro B are tocopherols and have been proven to show antioxidant properties (Bano and Deora, 2019). Thus, ATA is a compound of the alpha tocopherol group. The alpha tocopherol (vitamin E) is identified in Andrographis paniculata crude extract (Faisal et al., 2021), which is one of the ingredients of PHE in the present study. Alpha-tocopherol content is also reported in different parts of the Andrographis paniculata (Anuradha et al., 2010) plant in several other studies. Based on the results of a GC–MS study that was conducted on extracts of Andrographis paniculata that had been made public in the past, it has been hypothesized that ATA may play a significant role in a wide variety of diseases. Numerous studies have all come to the same conclusion, which is that ATA is a therapeutic component of the extracts that they investigated. Some examples of these studies were included in our earlier report (Singh et al., 2023).
Before the network pharmacology study, ADMET screening (Table 1) along with adverse drug reaction prediction (Table S1), and drug likeness score (Table 1) for all compounds of PHE, was found to be good for biological activity evaluation (Khanal and Patil, 2021). The compound ATA from PHE has also generated an excellent score and has been selected for the present study based on the above prediction data as well as its therapeutic importance in a variety of diseases as documented in the literature. In this study, system biology tools as well as network pharmacology tools were used, such as compound protein targets, interactions of protein with protein, in silico molecular docking, protein-pathway interactions, as well as interactions of different pathways (Khanal et al., 2019; Lee et al., 2018; Zhang et al., 2019). In addition, gene set enrichment analysis aids in the identification of genes and proteins that are involved in the pathophysiological processes (Subramanian et al., 2005) of DM. Throughout the network pharmacology study, the node (target, pathway, or phytoconstituent) with the maximum edge number is a highly anticipated molecule or a target or pathway that is highly regulated (Khanal and Patil, 2021). In addition to being a well-known cytokine, “TNF-α” that regulates synthesis, metabolism, and protein functions, it also has a strong association with diabetes (Khanal and Patil, 2021; Lu et al., 2022). This study found that the PHE has common intersecting genes such as AKT1 and TNF-α with the highest degree value among others, and the gene TNF-α was also the top intersecting gene for ATA. The docking study also predicted that ATA would have the maximum binding affinity for AKT1 and TNF-α, which are thought to be responsible for maintaining β-islet morphometry, insulin secretion regulation, and cell proliferation (Cruz et al., 2013; Khanal and Patil, 2021; Lu et al., 2022; Zhao et al., 2019).
Functional annotation from the GO study for selected hub genes created valuable and valid insights in the present study. Certain molecular functions (Fig. 2D) identified in the GO study, such as cysteine-type endopeptidase activity involved in apoptotic signaling pathways and processes, MAP kinase activity, and protein serine and threonine kinase activities, were found to be important in the overall management of diabetes at the gene level. Cellular responses to abiotic and environmental stimuli, chemical stress, and oxidative stress were some of the specifically identified biological processes (Fig. 2E) in the GO study against DM. Some cyclins, like A1, E2, and E1 complexes with CDK2, were involved in the protein serin/threonine kinase activities that regulate insulin receptor signaling pathways (IRS) as an acellular component (Fig. 2F) in the GO study. In addition, mitochondria, nucleoplasm, nuclear lumen, and others were also involved as prime cellular components in the study. The KEGG pathway gene enrichment analysis for optimized genes in this study dictated the important mechanism for management of the highly referred metabolic disorder (DM). In this scenario, the optimized important signaling pathways presented a new insight into managing DM with the essential active compound ATA from PHE. So, ATA stimulates tyrosine phosphorylation of the IRS β-subunit by insulin binding, resulting in the stimulation of two significant signaling pathways: PI3K/Akt and MAPK. As a result, a signaling cascade is set off, triggering a variety of biological responses. Stimulating insulin receptors has both short-term and long-term effects. For example, the glucose transporter GLUT4 moves to the surface of target cells, the expression of glucokinase goes up, and the expression of gluconeogenic as well as ketogenic enzymes produced by the liver goes down (downstream effects). So, we predicted that ATA stimulates the PI3K-AKT (phosphatidylinositol 3-kinase-Akt) pathway, which is solely accountable for glucose metabolism and inhibition of gluconeogenesis. The MAPK pathway is responsible for gene expression and also interacts with the PI3K-Akt (KEGG: hsa04151) pathway to regulate differentiation and proliferation of the β-subunit of IR in the complete pathogenesis of DM in earlier literature (Cruz et al., 2013; Lu et al., 2022; Song et al., 2018; Mukherjee et al., 2021; Zhao et al., 2019), which was consistent with our prediction.
TNF-α, interferon-alpha, interleukin-1, and interleukin-6 have all been reported to significantly regulate metabolism, synthesis, protein function, and other cytokine functions. According to the findings of the current network pharmacology study, the bioactive components of PHE were also reported to regulate six genes (MAPK1, MAPK14, MAPK8, CASP3, CASP8, and TNF) through the regulation of cytokine-cytokine interactions (KEGG: hsa04668). The significant reduction in urea, creatinine, and uric acid levels may be the consequence of cytokine regulation or perhaps the modulation of PI3K-Akt-mediated gluconeogenesis, among many other things (Khanal and Patil, 2021; Lu et al., 2022). Thus, the entire KEGG pathway enrichment study provides extensive genetic information about diabetes management. All the pathways, such as anti-inflammatory pathways, immune responsive pathways, concurrent disease-related pathways, etc., dictated in this study are important pathways for cumulative and complete management of DM. Further, biological activity spectra (PASS) prediction for the ATA (Fig. 3B) like hypolipemic, retinoprotector, antihypercholesterolemic, cholesterol synthesis inhibitor, etc. with a versatile nature produces a green insight for the management of metabolic disorders (DM; Table S5). Furthermore, higher affinity binding scores of ATA, along with strong binding types and binding residues obtained (Table S6) for the crucial and selected hub genes (AKT1 and TNF), were observed to be effective in DM (Zhao et al., 2019). Ramchandran plots (Supplementary Figs. S3 and S4) and conformational quality evaluations (Supplementary Figs. S5 and S6) are also important for the structural validation of receptor protein-related hub genes in the study (Balogun et al., 2022). The investigation of docked conformational stability is an essential step in determining and understanding the process of protein inhibition prophesied by the autodock method. The higher binding affinity of both hub genes to docked ATA complexes (Supplementary Table S6), as well as the docked conformational poses of ATA (Fig. 3C–F) and Supplementary Figs. S7 and S8, was intriguing for further studies. According to the findings of the MD simulation study, the MD simulation trajectory gives a deeper understanding of the conformational scenery of a docked protein–ligand complex under specific conditions such as temperature and pressure, which are dependent on the simulation threshold. The formation of intermolecular contacts between ligands and amino acids in the active site of the protein indicates that the compound forms at least one hydrogen bond with the amino acid residues inside the active site of the protein. It is primarily used to compute the average distance for the simulated protein–ligand complex by RMSD. During the 100 ns simulation period, the Cα atoms in the AKT1 complex docked with compound ATA and the Cα atoms in TNF-α docked with compound ATA both showed a relatively stable RMSD value, which is acceptable for small globular proteins. The RMSD of a protein-fit ligand is a way of measuring just how stable the ligand is still within the protein's selective binding pocket. After being identified as having significant structural stability in the preferential pocket of AKT1 as well as TNF-α, the ATA bioactive compound derived from PHE was also demonstrated to have equivalent fluctuations (RMSF) for improved stability. For the first time, during the 100 ns simulation process, it was observed that the docked ligand formed strong interactions with the amino acid residues (seen in protein ligand contact mapping) in the selective pocket of both the AKT1 and TNF-α crystal structures. In general, these findings show that the PHE ligand (ATA) had a similar binding orientation to AKT1 and TNF-α, and it also had a lot of stability in the preferred pocket of AKT1 and TNF-α (Khanal and Patil, 2021).
MM/GBSA is a standard method for structure-based drug design because of its efficiency, availability of free software, and use of standard scoring features to measure the binding energy of a protein–ligand complex, mostly under the docking model. Docking model initiatives, on the other hand, are deficient in important energy parameters like solvation energy systems in the measurement of bonding free energy. As a result, the binding free energy obtained through docking methods is not highly accurate, and it is difficult to distinguish between docked poses that have similar binding configurations and affinities. This threat has been addressed by the development of the MM/GBSA algorithm, which has been shown to be reliable and accurate in computing the strength and rigidity as well as the binding free energy for protein–ligand complexes (Sakkiah et al., 2021). Furthermore, the results of the MM/GBSA test (the higher values of MM/GBSA docked complexes) recommended that the ATA be a new therapeutic applicant with the highest AKT1 and TNF-α active site saturation as well as the most favorable binding alignment. As a result, the PHE compound ATA can also be recommended as a potent and selective inhibitor of AKT1 and TNF-α.
Many studies have pointed to the potential role that medicinal plant extracts can play as α-glucosidase inhibitors, indicating that the ability of extracts to handle hyperglycemia is a real possibility. The presence of bioactive compounds in the extract can be credited with the activity that it possesses (Oh et al., 2021). In the present study, PHE explicated a remarkable inhibition potential (Supplementary Table S8) against the α-glucosidase enzyme. Moreover, synergistic good antioxidant values with enrichment of medicinal compounds were discovered in PHE, making it a potent anti-inflammatory drug (Singh et al., 2023), corroborating our earlier statement. Therefore, PHE containing bioactive compounds was found to be a significant and synergistically effective antidiabetic drug for the treatment of DM. As a result, we came to the conclusion that our research demonstrated the potent α-glucosidase inhibitory potential of PHE and suggested that the activity was due to the synergistic potential of bioactive constituents present in it. This also verified the present network pharmacology study of the bioactive compound ATA present in PHE. In conclusion, we found that our research demonstrated the potent α-glucosidase inhibitory potential of PHE.
5 Conclusion
Initially, a network pharmacology strategy was used to examine PHE for its bioactive ingredients and mechanisms in relation to diabetic patients. The potential activity of PHE against diabetes was found to involve the compounds and common genes in the network. The most important finding, however, was that PHE is effective in warding off diabetes and that ATA is a key active component and TNF is a key hub gene. The in-vitro anti-diabetic activity confirmed that the key signaling pathway enriched with the hub gene also stimulates signaling pathways like MAPK and PI3K/Akt, which may enhance insulin receptor (IR) autophosphorylation. Overall, this study provides pharmaceutical justification for further interpretations of the bioactive components and mechanisms of PHE against DM as well as scientific validation of the therapeutic efficacy of the ATA of PHE on diabetes through in vivo pharmacological activity.
CRediT authorship contribution statement
Amit Kumar Singh: Conceptualization, Funding acquisition, Data curation, Writing – original draft, Writing – review & editing, Investigation, Methodology. Pradeep Kumar: Funding acquisition, Data curation, Writing – review & editing, Investigation. Sunil Kumar Mishra: Conceptualization, Validation, Formal analysis, Methodology, Supervision. KavindraNath Tiwari: Conceptualization, Resources. Anand Kumar Singh: Writing – review & editing, Visualization, Software. Ajay Kumar Pandey: Visualization. Ali A. Shati: Validation. Mohammad Y. Alfaifi: Formal analysis. SeragEldin I. Elbehairi: Formal analysis. R.Z. Sayyed: Formal analysis.
Acknowledgements
The authors sincerely appreciate the Deanship of Scientific Research at King Khalid University in Abha, Saudi Arabia, provided funding for this search through large groups (Project under Grand number R.G.P. 1/430/44). The author Amit Kumar Singh is thankful to the Indian Institute of Technology (BHU) and MHRD, India, for fellowship. Author Kavindra Nath Tiwari thankfully acknowledges Banaras Hindu University, Varanasi for providing financial assistance through an incentive grant under the Institutions of Eminence (IoE) scheme 6031 for research.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- The management of diabetes mellitus-imperative role of natural products against dipeptidyl peptidase-4, α-glucosidase and sodium-dependent glucose co-transporter 2 (SGLT2) Bioorg. Chem.. 2019;86:305-315.
- [Google Scholar]
- Network pharmacology-based anti-pancreatic cancer potential of kaempferol and catechin of Tremaorientalis L. through computational approach. Med. Oncol.. 2023;40(5):133.
- [Google Scholar]
- Enzymes inhibitors from natural sources with antidiabetic activity: a review. Phytother. Res.. 2019;33(1):41-54.
- [Google Scholar]
- Plant growth regulators induced changes in antioxidant potential and andrographolide content in Andrographis paniculata Wall. exNees. Pestic. Biochem. Phys.. 2010;98:312-316.
- [Google Scholar]
- Discovery of potential HER2 inhibitors from Mangiferaindica for the treatment of HER2-Positive breast cancer: an integrated computational approach. J. Biomol. Struct. Dyn.. 2022;40(23):12772-12784.
- [Google Scholar]
- Preliminary phytochemical screening and GC-MS analysis of methanolic leaf extract of Abutilon pannosum (Forst. F.) Schlect. from Indian Thar desert. J. Pharmacogn Phytochem.. 2019;8(1):894-899.
- [Google Scholar]
- In vitro evaluation of the α-glucosidase inhibitory potential of methanolic extracts of traditionally used antidiabetic plants. BMC Complement. Altern. Med.. 2019;19:74.
- [Google Scholar]
- Current research trends in traditional chinese medicine formula: a bibliometric review from 2000 to 2016. Evid. Based Complement Alternat. Med.. 2019;2019:3961395.
- [Google Scholar]
- The linkage between inflammation and Type 2 diabetes mellitus. Diabetes Res. Clin. Pract.. 2013;99(2):85-92.
- [Google Scholar]
- Dichloromethane crude extract of Gymnanthemumextensumcombined with low piperine fractional Piper nigrumextract induces apoptosis on human breast cancer cells. Indian J. Pharm. Sci.. 2021;83(2):247-260.
- [Google Scholar]
- Prediction of the biological activity spectra of organic compounds using the PASS online web resource. Chem. Heterocycl. Compd.. 2014;50(3):444-457.
- [Google Scholar]
- Network-based approaches to explore complex biological systems towards network medicine. Genes. 2018;9(9):437.
- [Google Scholar]
- Salix eleagnosScop. – a novel source of antioxidant and anti-inflammatory compounds: biochemical screening and in silico approaches. S. Afr. J. Bot.. 2020;128:339-348.
- [Google Scholar]
- IDF Diabetes Atlas 2022 reports (10th ed.). International Diabetes Federation; 2022.
- Current anti-diabetic agents and their molecular targets: a review. Eur. J. Med. Chem.. 2018;152:436-488.
- [Google Scholar]
- Integration of network and experimental pharmacology to decipher the antidiabetic action of Durantarepens L. J. Integr. Med.. 2021;19(1):66-77.
- [Google Scholar]
- In silico docking study of limonoids from Azadirachtaindica with pfpk5: a novel target for Plasmodium falciparum. Indian J. Pharm. Sci.. 2019;81:326-332.
- [Google Scholar]
- Cardiovascular safety of anti-diabetic drugs. Eur. Heart. J. Cardiovasc. Pharmacother.. 2016;2(1):32-43.
- [Google Scholar]
- Identification and validation of core genes as promising diagnostic signature in hepatocellular carcinoma based on integrated bioinformatics approach. Sci. Rep.. 2022;12(1):19072.
- [Google Scholar]
- Network pharmacology-based study on apigenin present in the methanolic fraction of leaves extract of Cestrum nocturnum L. to uncover mechanism of action on hepatocellular carcinoma. Med. Oncol.. 2022;39(10):155.
- [Google Scholar]
- Network pharmacology-based prediction of active compounds and molecular targets in Yijin-Tang acting on hyperlipidaemia and atherosclerosis. J. Ethnopharmacol.. 2018;221:151-159.
- [Google Scholar]
- Tumor necrosis factor-α signaling in nonalcoholic steatohepatitis and targeted therapies. J G. G = Yi Chuanxuebao. 2022;49(4):269-278.
- [Google Scholar]
- Network pharmacology in research of chinese medicine formula: methodology, application and prospective. Chin. J. Integr. Med.. 2020;26(1):72-80.
- [Google Scholar]
- Molecular combination networks in medicinal plants: understanding synergy by network pharmacology in Indian traditional medicine. Phytochem. Rev.. 2021;20:693-703.
- [Google Scholar]
- Active ingredients and mechanisms of Phellinuslinteus (grown on Rosa multiflora) for alleviation of Type 2 diabetes mellitus through network pharmacology. Gene. 2021;768:145320
- [Google Scholar]
- Network pharmacology of cancer: from understanding of complex interactomes to the design of multi-target specific therapeutics from nature. Pharmacol. Res.. 2016;111:290-302.
- [Google Scholar]
- The potential of natural products for targeting PPARα. Acta Pharm. Sin. B. 2017;7(4):427-438.
- [Google Scholar]
- Computational assessment of the pharmacological profiles of degradation products of Chitosan. Front. Bioeng. Biotechnol.. 2019;7:214.
- [Google Scholar]
- The P4 health spectrum – a predictive, preventive, personalized and participatory continuum for promoting healthspan. Prog. Cardiovasc. Dis.. 2017;59(5):506-521.
- [Google Scholar]
- Elucidation of agonist and antagonist dynamic binding patterns in ER-α by integration of molecular docking, molecular dynamics simulations and quantum mechanical calculations. Int. J. Mol. Sci.. 2021;22(17):9371.
- [Google Scholar]
- Exploring patient perspectives of prediabetes and diabetes severity: a qualitative study. Psychol. Health. 2019;34(11):1314-1327.
- [Google Scholar]
- Phytochemicals, antioxidant, anti-inflammatory studies, and identification of bioactive compounds using GC-MS of ethanolic novel polyherbal extract. Appl. Biochem. Biotechnol. 2023 Advance online publication
- [CrossRef] [Google Scholar]
- AQR is a novel type 2 diabetes-associated gene that regulates signaling pathways critical for glucose metabolism. J. g. G = Yi Chuanxuebao. 2018;45(2):111-120.
- [Google Scholar]
- Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A.. 2005;102(43):15545-15550.
- [Google Scholar]
- Natural activators of adenosine 5'-monophosphate (AMP)-activated protein kinase (AMPK) and their pharmacological activities. Food Chem. Toxicol.. 2018;122:69-79.
- [Google Scholar]
- Natural products for the treatment of type 2 diabetes mellitus: pharmacology and mechanisms. Pharmacol. Res.. 2018;130:451-465.
- [Google Scholar]
- Lupenone is a good anti-inflammatory compound based on the network pharmacology. Mol. Divers.. 2020;24(1):21-30.
- [Google Scholar]
- Cyclocaryapaliurus leaves tea improves dyslipidemia in diabetic mice: a lipidomics-based network pharmacology study. Front. Pharmacol.. 2018;9:973.
- [Google Scholar]
- Network pharmacology databases for traditional chinese medicine: review and assessment. Front. Pharmacol.. 2019;10:123.
- [Google Scholar]
- Regulation of glucose metabolism by bioactive phytochemicals for the management of type 2 diabetes mellitus. Crit. Rev. Food Sci. Nutr.. 2019;59(6):830-847.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2024.103138.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




