

Translate this page into:
In silico exploration of potential PRKG1 Inhibitors: A comprehensive study using MTIOPEN Screening, molecular Docking, and MD simulation
⁎Corresponding author. aralonazi@ksu.edu.sa (Abdullah R. Alanzi)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
The enzyme protein kinase G1 (PRKG1) plays a crucial role in cellular signaling pathways such as smooth muscle relaxation, neuronal signaling, and platelet aggregation. The dysregulation of PRKG1 leads to different diseases, making it a promising drug target. In this study, we employed a comprehensive in silico strategy to explore potential inhibitors of PRKG1 by using the crystal structure of the PRKG1 protein. The active site of PRKG1 protein was parameterized within a three-dimensional grid box. The 100 hit compounds identified during virtual screening were docked to the prepared PRKG1 receptor to predict binding affinities. The top ten compounds were chosen, and their binding affinities ranged from −10.734 to −10.398 kcal/mol. Finally, a 200 ns long simulation was run to confirm the stability of the protein–ligand complexes at the binding sites of the two top compounds against the PRKG1 receptor. All these findings suggest that the selected compounds can serve as potential compounds to inhibit the PRKG1 function in biological assays.
Keywords
Protein Kinase G1 (PRKG1)
Cellular Signaling Pathways
Virtual Screening
Inhibitors
Molecular Dynamics (MD) Simulation

1 Introduction
Protein Kinase G1 (PRKG1) is a key enzyme in the family of cGMP-dependent protein kinases. It is critical for mediating cellular responses to the second messenger cGMP, which is a key signaling molecule involved in many physiological processes. Many cellular activities are regulated by the serine/threonine kinase PRKG1, which phosphorylates target proteins in response to intracellular cGMP levels (Schall et al., 2020, Khiro and Hasan 2023).
PRKG1 has a modular structure that consists of several functional domains. The key domains are an N-terminal regulatory domain with two tandem cGMP-binding domains (CNB-A and CNB-B), a hinge region, a catalytic domain that is responsible for kinase activity, and a C-terminal autoinhibitory domain. The presence of these domains allows PRKG1 to change conformation in response to cGMP binding, resulting in its activation (Browning et al., 2010).
The activation of PRKG1 is intricately linked to cGMP levels within the cell. This activation mechanism is essential for PRKG1′s function as a cGMP signaling mediator. Numerous physiological processes involving various tissues and cell types are regulated by PRKG1. It controls synaptic transmission, platelet aggregation, vascular tone, and smooth muscle relaxation. PRKG1 activation in vascular smooth muscle cells, for example, causes vasodilation by phosphorylating target proteins involved in smooth muscle cell relaxation (Norton et al., 2019, Henning 2022).
PRKG1 dysregulation has been linked to a variety of disease states. Variations in PRKG1 activity have been connected to high blood pressure and atherosclerosis, two cardiovascular disorders. Additionally, PRKG1 dysfunction has been linked to neurological disorders such as migraine and neurodegenerative diseases. Understanding the role of PRKG1 in disease pathogenesis opens up possibilities for therapeutic intervention (Gago‐Díaz et al., 2016, Henning 2022).
Given its critical role in a variety of physiological and pathological processes, PRKG1 has emerged as a promising drug target. Identification of small molecules that modulate PRKG1 activity can provide potential therapeutic strategies for diseases associated with aberrant cGMP signaling. Hence, this study has been designed to identify potential inhibitors against PRKG1 protein utilizing molecular modelling and simulation techniques.
2 Methodology
2.1 Virtual screening
The structure-based virtual screening is used to identify small number of compounds from the libraries of small molecules containing millions of compounds. For structure-based virtual screening, 3D grid information is required to inform the program exactly what space to look for.
2.2 3D grid parametrization
Using Autodock Tools, a 3D grid box containing the protein active site (PDB ID: 3OCP) was generated to determine the coordinates in order to create a customized 3D environment for structure-based virtual screening (Pacheco and Hpc 2012).
2.3 Virtual screening using MTiOpenScreen
The structure-based virtual screening of PRKG1 protein for was performed using MTiOpenScreen (Dwivedi et al., 2021). Among the significant libraries of classified compounds that MTiOpenScreen provides are collections of natural product compounds (NP-lib), pharmaceuticals-lib, which is a collection of purchasable chemicals, and FOOD-lib, which is a collection of food component compounds. Autodock Vina is automated by this server for virtual screening (Trott and Olson, 2010). The PRKG1 protein target was the target of a virtual screening using the drugs library to identify hit compounds.
2.4 Molecular docking
The 3D structure of PRKG1 protein was prepared using Maestro's protein preparation wizard (Madhavi Sastry et al., 2013). The protein preparation involved three steps; preprocessing that deals with the addition of hydrogens to protein, removing the crystal water molecules, addition of charges and repairing of side chain residues. In next step, the hydrogens were optimized and different tautomeric states were produced at pH 7.0 by employing PROPKA (Sadeer et al., 2019). Lastly, the optimized structure was minimized by using the OPLS_2005 forcefield (Shivakumar et al., 2012). The natural substrate was used to conduct site-specific docking, which generated the grid. The coordinates for X, Y, and Z have values of −0.08, 36.96, and 26.83. After grid generation, the ligands were prepared using LigPrep (Matsuoka et al., 2017). The different ionization states of the ligands were generated by using Epik at pH 7. Similarly, several stereoisomers of ligands were generated by employing the OPLS_2005 forcefield. Finally, the prepared ligands were docked to the PRKG1 receptor by using the glide tool.
2.5 MD simulation
Desmond was employed to conduct Molecular Dynamics simulations lasting 200 ns for selected compounds to find the behavior of complex during simulation (Bowers et al., 2006). The stability of complexes was assessed through MD simulations, following a series of steps, including preprocessing, optimizing the hydrogen atoms, and by minimizing the complex by utilizing the OPLS_2005 force field (Shivakumar et al., 2012). The protein–ligand complexes were solvated in TIP3P water box of 10 Å (Price and Brooks III, 2004). The systems were neutralized by the addition of Na+ and Cl- counter ions and 0.15 M NaCl salt to mimic physiological conditions. The prepared systems were subjected to production run by using the NPT ensemble at 300 K temperature and 1 atm pressure. During production, the MD trajectories were recorded at 40 ps intervals to analyze the results by Simulation Interaction Diagram module.
2.6 MMGBSA
The free energy of the selected complexes was calculated by employing the Prime-MMGBSA module of Schrödinger (Prema et al.,). The presence of the counter ions in the system was stripped and the VSGB solvent model along with OPLS_2005 forcefield were employed to calculate the binding free energy (ΔGbind). The calculations were conducted by using Equation (1). The binding free energy is the difference between complex free energy and the free energy of protein and ligand. The free energy terms used during the calculation were ΔGcoulomb, ΔGcovalent, ΔGHbond, ΔGLipo, ΔGPacking, ΔGvdW, ΔGstraing_energy, and ΔGSolv_Gb.
3 Results
3.1 3D grid parametrization
The residues of the PRKG1 protein's binding pocket were parameterized inside a three-dimensional grid box. The crystal ligand was used to measure the 3D coordinates. The cartesian coordinates for X, Y, and Z were −0.08, 36.96, and 26.83, respectively. The visual representation of 3D grid box is shown in Fig. 1.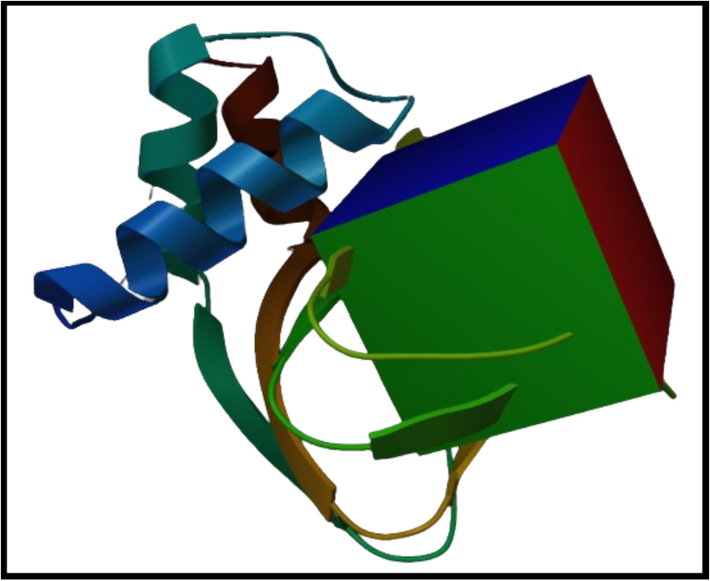
The representation of grid box around the binding pocket of PRKG1.
3.2 Virtual screening of PRKG1 in MTiOpenScreen
MTiOpenScreen was employed to perform the virtual screening of the PRKG1 protein using Drug-lib including 21,276 compounds based on binding affinities (Kumar and World, 2019). A total of 100 compounds were evaluated against PRKG1.
3.3 Molecular docking
The 100 screened hits were prepared by LigPrep and the docked to the PRKG1 receptor to find the binding affinities of the compounds. The performance of the compounds was evaluated by measuring the binding affinities and the ten compounds with best docking scores were selected for further analysis (Table 1). The docking scores of the selected hits indicated that these have the probability of inhibiting the function of PRKG1 protein.
Sr.
Compounds
Structures
Glide score (kcal/mol)
1
Orvepitant
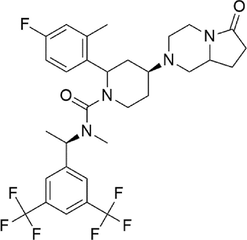
−8.691
2
Fosaprepitant
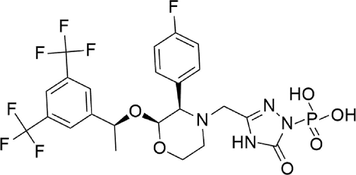
−8.183
3
Oxalinast
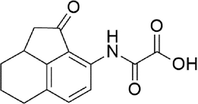
−8.127
4
Pretomanid
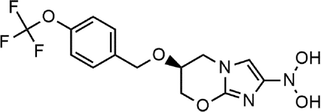
−8.094
5
Ipsalazide
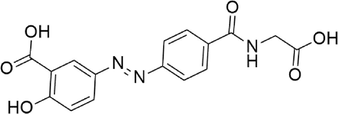
−7.944
6
Polydatin
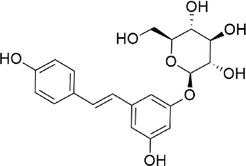
−7.888
7
Delamanid
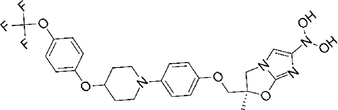
−7.867
8
Edotecarin
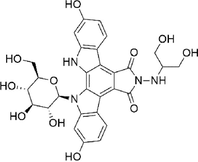
−7.765
9
Imanixil
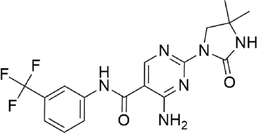
−7.636
10
Salazosulfamide
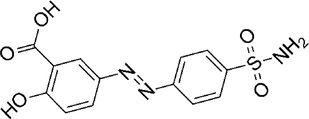
−7.584
3.4 Molecular interactions analysis
The molecular interactions of the selected hits with the binding pocket of PRKG1 receptor were analyzed using the Discovery Studio client tool. The observed interactions involved: conventional hydrogen bond, carbon hydrogen bond, van der Waal interactions, Pi-Sulfur, Amide Pi-Stacked, Halogen, and Alkyl interactions. These interactions play a pivotal role in determining the binding affinities and docking scores for each of the top candidate compounds. Notably, the formation of intermolecular hydrogen bonds between the ligand and the amino acid within the active sites has a significant impact on the overall strength of the resulting complex. Consequently, these interactions consistently enhance the docking results (Thillainayagam et al., 2018). Orvepitant formed two conventional hydrogen bonds with Asp117, Thr193, two carbon hydrogen bonds with Asn116, Glu183, and seven alkyl interactions with Phe118, Lys179, Met175, Val165, Leu184, Cys173, Leu172 (Fig. 2A). Fosaprepitant formed three conventional hydrogen bonds with Asp115, Asp117, Val180, two carbon hydrogen bonds with Glu183, Asn116, one Pi-Sigma interaction with Leu172, and four alkyl interactions with Ala194, Val165, Leu184, Lys179 (Fig. 2B). Similarly, Oxalinast made three conventional hydrogen bonds with Gly182, Thr193, Ala194, one carbon hydrogen bond with Arg192, and three alkyl interactions with Val165, Cys173, Met175 (Fig. 2C). Lastly, Pretomanid made two conventional hydrogen bonds with Asp117, Thr193, one carbon hydrogen bond with Asn116, one halogen interaction with Asp115, and two salt bridges with Glu183, Arg192 (Fig. 2D). The molecular interactions of other compounds are shown in Table 2.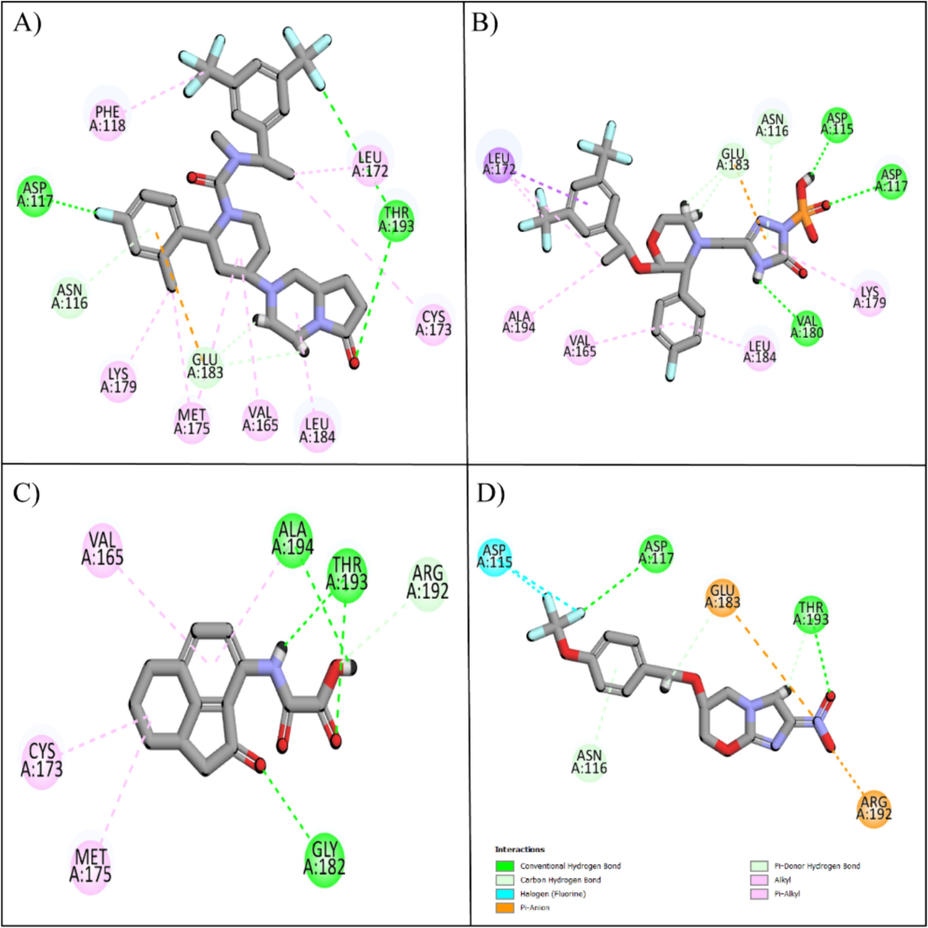
The hit and control compounds' molecular interactions. A) Orvepitant, B) Fosaprepitant, C) Oxalinast, and D) Pretomanid. Green spheres represent hydrogen bonds, magenta spheres represent hydrophobic interactions, orange spheres represent salt bridges, and cyan spheres represent halogen interactions.
Sr.
Compound code
Interactions
1
Orvepitant
Conventional Hydrogen Bond: Asp117, Thr193
Carbon Hydrogen Bond: Asn116, Glu183
Alkyl: Phe118, Lys179, Met175, Val165, Leu184, Cys173, Leu172
2
Fosaprepitant
Conventional Hydrogen Bond: Asp115, Asp117, Val180
Carbon Hydrogen Bond: Glu183, Asn116
Pi-Sigma: Leu172
Alkyl: Ala194, Val165, Leu184, Lys179
3
Oxalinast
Conventional Hydrogen Bond: Gly182, Thr193, Ala194
Carbon Hydrogen Bond: Arg192
Alkyl: Val165, Cys173, Met175
4
Pretomanid
Conventional Hydrogen Bond: Asp117, Thr193
Carbon Hydrogen Bond: Asn116
Salt Bridge: Glu183, Arg192
Halogen: Asp115
5
Ipsalazide
Conventional Hydrogen Bond: Asp115, Glu183, Ala185, Thr193, Ala194
Carbon Hydrogen Bond: Asp117, Gly182
Alkyl: Leu184
6
Polydatin
Conventional Hydrogen Bond: Ala194, Thr193, Cys173, Asn116, Asp115, Asp117
Carbon Hydrogen Bond: Arg192
Alkyl: Ile146, Leu184
7
Delamanid
Conventional Hydrogen Bond: Cys190, Thr193, Gly182
Pi-Sigma: Leu172
Alkyl: Tyr188, Leu184, Val165, Ala194, Met175, Lys167
8
Edotecarin
Conventional Hydrogen Bond: Lys179, Asp117, Asp115, Glu183, Thr193
Alkyl: Met175, Val165, Cys173, Leu172
9
Imanixil
Van der Waal: Gly101
Conventional Hydrogen Bond: Ala102, Asp232, Lys121
Pi-Cation: Met169
Alkyl: Arg106, Ala231, Val106, Ala119, Leu221, Phe384, Ile98, Tyr171
10
Salazosulfamide
Conventional Hydrogen Bond: Asp117, Glu183, Ala185, Arg192, Thr193, Ala194
Pi-Anion: Met175
Alkyl: Lys179, Leu184
3.5 Binding pose analysis
After the molecular interaction analysis, the plausible binding poses of the top two hits were analyzed by aligning them on the crystal ligand. The analysis showed that the docked compounds showed perfect alignment on the co-crystal ligand with similar binding mode (Fig. 3). Thus, the binding poses of the selected compounds were subjected towards the stability analysis by employing the MD Simulation study.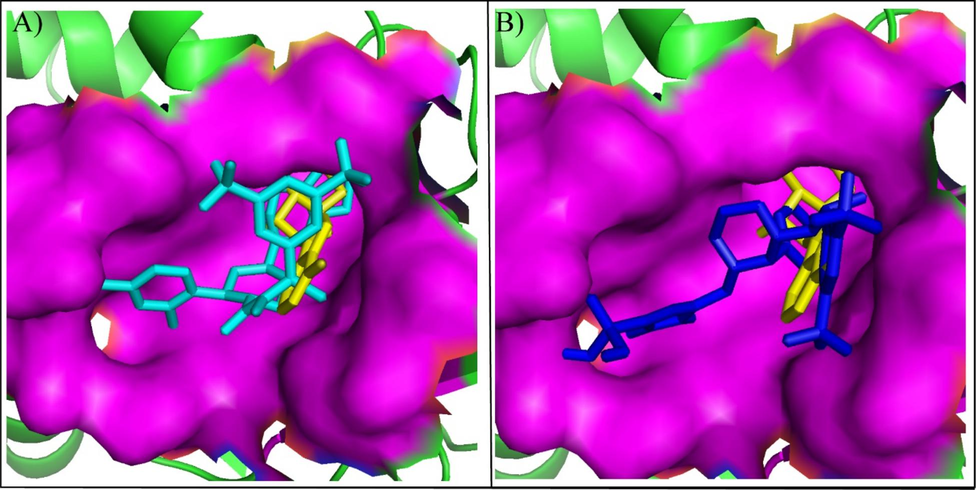
The alignment of binding modes of the two compounds with co-crystal ligand (Yellow sticks). (A) Orvepitant (Cyan sticks), (B) Fosaprepitant (Blue sticks).
3.6 MD simulation
3.6.1 RMSD
The protein–ligand complexes were subjected to the 200 ns simulation to evaluate the stability of the complexes and find the fluctuations in the protein region in the presence of docked ligands. The RMSD of carbon alpha (C) atoms was calculated to find the overall structural alterations and deviations of the complexes during the simulation (Sargsyan et al., 2017). The Orvepitant complex's RMSD values remained in the 2 Å range until 140 ns, when they gradually increased to 3 Å at 150 ns, and they remained in this range until end of simulation. On the other hand, the RMSD of ligand fit was aligned on the protein (Fig. 4A). The RMSD of fosaprepitant first varied by up to 4 Å over the first 50 ns, then at the 60 ns point, it stabilized in the 3 Å range. After 60 ns, it showed no change and remained constant during the simulation, while the RMSD of ligand was lower than protein during 50 to 150 ns but it aligned on protein in last phase of simulation (Fig. 4B). The deviations in the RMSD values were due to the presence of loop region at the N terminal of the protein. The flexibility of the loop caused major deviations in the carbon alpha atoms of the protein during simulation.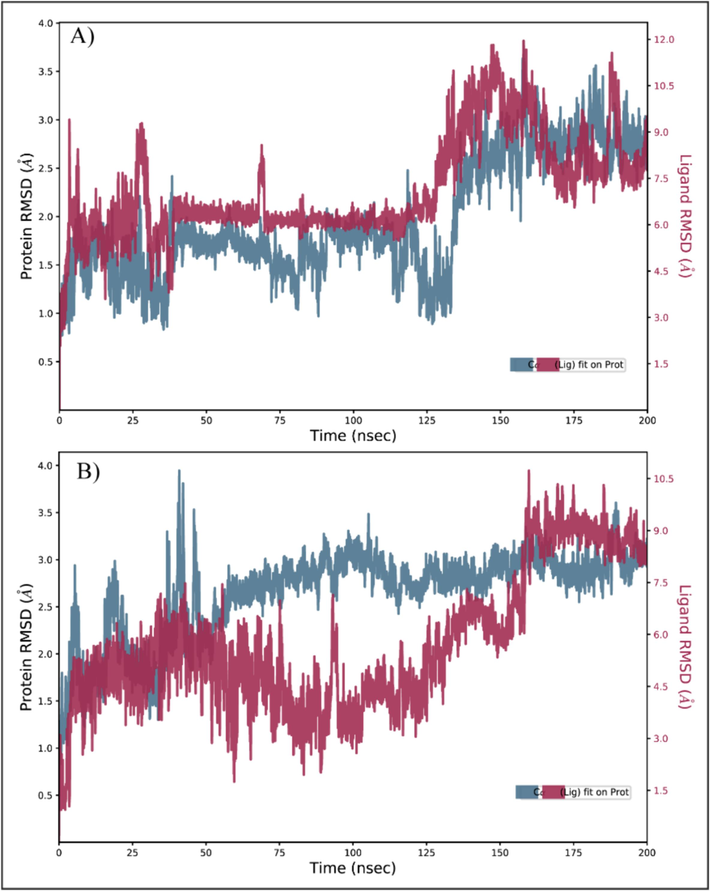
The RMSD of carbon alpha atoms of PRKG1 in complex with the ligands during 200 ns simulation. (A) Orvepitant, (B) Fosaprepitant.
3.6.2 RMSF
RMSF analysis has been conducted to determine the fluctuation of the proteins while they are bound to the ligands (Martínez, 2015). For each protein residue over the simulation period, RMSF values give detailed information on the residue's mobility and flexibility. Based on the expected RMSF values, most protein residues changed very slightly during the simulation, which was less than 2 Å. This suggests that these residues maintained their relative stability and stiffness while the ligands were present. However, the RMSF values in the protein's loop region i.e., residues ranging from 1 to 24 were higher with values reaching 9 Å (Fig. 5). According to the RMSF analysis, most of the residues in the protein and ligand complexes held onto their rigid forms, causing the complex to stay stable. RMSF values of loop regions were higher, indicating that these areas displayed larger variations and could have engaged in dynamic interactions with the ligands. Most protein residues showed slight changes, but loop parts showed significantly higher levels of flexibility. Overall, the notion of stable protein and ligand complexes is consistent with RMSF values.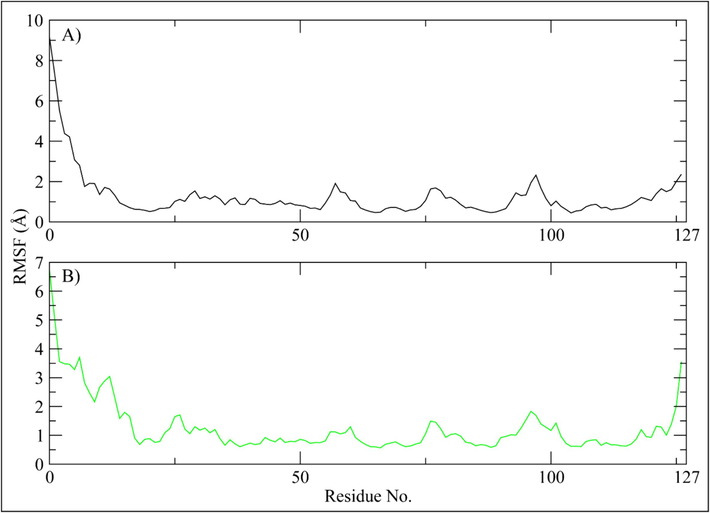
The residual fluctuations of the PRKG1 receptor upon binding of the selected compounds. (A) Orvepitant, (B) Fosaprepitant.
3.6.3 Protein-Ligand contacts
The Simulation interaction diagram showed that the protein–ligand contacts deal with hydrogen bonding, ionic bonds and hydrophobic interactions which are important for the stability of complex Residues that form hydrogen bonds with Orvepitant were Leu96, Cys173, Thr193, and Ala194 (Fig. 6A). In the Fosaprepitant complex, the residues involved in hydrogen bonding were Ala112, Asp115, Asn116, Asp117, Lys179, Val180, Glu183, Thr193, and Ala914 (Fig. 6B). These hydrogen bonding interactions, which were displayed during the MD simulations, not only highlighted the specific residues that were crucial for stabilizing the protein–ligand complexes, but they also provided insight into the crucial interactions that underpin the complexes' general stability and binding affinity.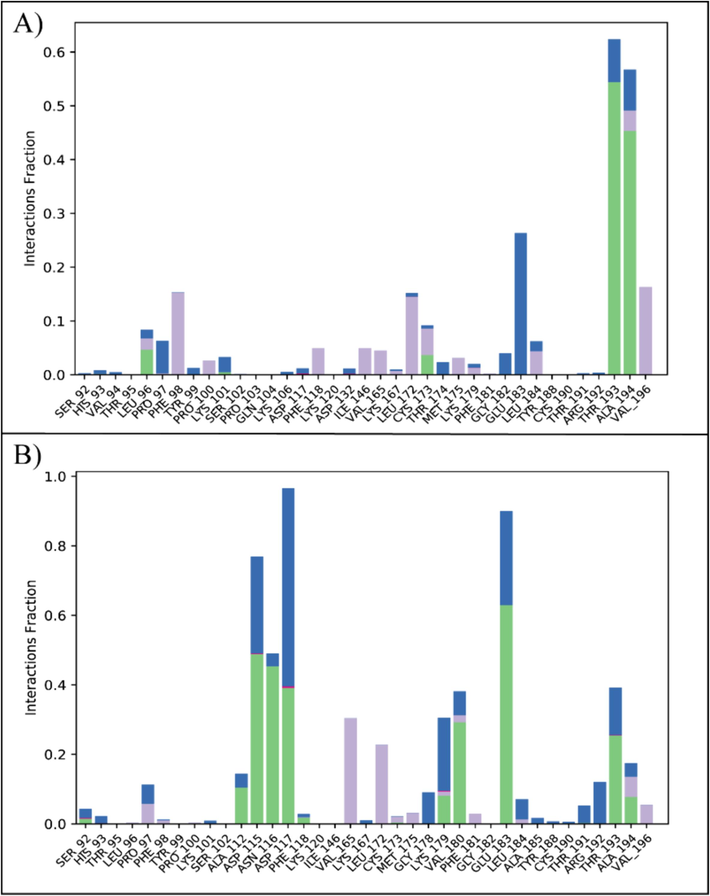
The protein–ligand contacts of PRKG1 complexes. (A) Orvepitant, (B) Fosaprepitant. Green bars show hydrogen bonding, gray shows the hydrophobic interactions, and blue shows the water bridges.
3.6.4 Hydrogen bonding
Hydrogen bonding plays a significant role in the protein–ligand complex stability. Hence, the number of hydrogen bonds between ligand and active site residues were calculated over the simulation. The hydrogen bonding plots indicate that Orvepitant made at least one hydrogen bond throughout the simulation. Some frames showed two hydrogen bonds while three bonds were also observed (Fig. 7A). Fosaprepitant made at least three hydrogen bonds in the first half while the hydrogen bonds decreased to 2 in the second half of simulation (Fig. 7B).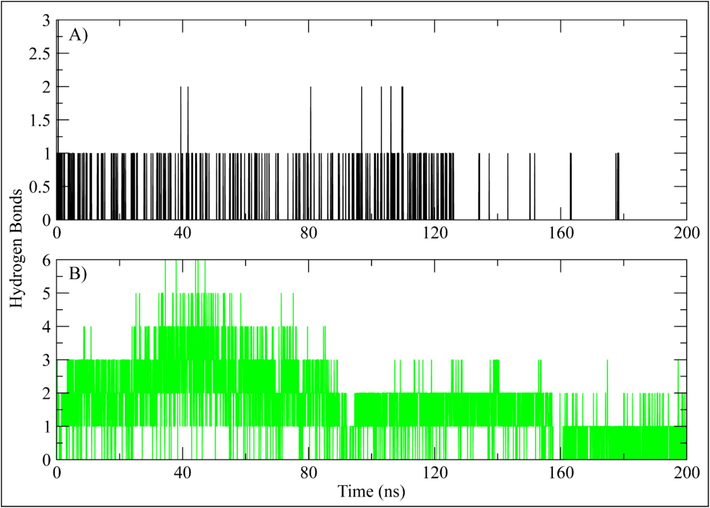
The number of hydrogen bonds between PRKG1 and selected ligand calculated during 200 ns simulation. (A) Orvepitant, (B) Fosaprepitant.
3.7 MM/GBSA
The prime-MMGBSA module was used to calculate the binding energy of selected complexes (Godschalk et al., 2013). The binding energies of the Orvepitant and Fosaprepitant complexes were −82.59, and −76.75 kcal/mol, respectively. Gbind resulted from non-bonded interactions, GCoulomb, GPacking, GHbond, GLipo, and GvdW (Table 3). GbindLipo, GbindvdW, and GbindCoulomb affected the average binding free energies among all interaction types. Conversely, the final average binding energies were least affected by the GbindSolvGB and GbindCovalent energies. Furthermore, stable hydrogen bonds were observed between the ligands and amino acid residues indicated by GbindHbond interaction values. Thus, the binding energies calculated during simulation supported the binding affinities of ligands obtained during docking studies (Decherchi and Cavalli 2020).
Orvepitant
Fosaprepitant
Bind
−82.592
−76.759
Coulomb
−15.7899
−24.1222
Covalent
10.70593
2.827797
Hbond
−0.6553
−1.12823
Lipo
−59.6839
−31.7532
Packing
−0.16392
−0.49296
Strain_Energy
21.70663
19.46657
Solv_GB
−38.7115
−41.5568
vdW
18.8572
6.631646
4 Discussion
PRKG1 is a multifunctional protein kinase that acts as a mediator in cGMP signaling pathways, influencing a variety of cellular processes. Its complex regulatory mechanisms and involvement in both physiological and pathological conditions make it a fascinating research topic with important implications for drug discovery and therapeutic interventions. The in-silico approach significantly accelerates the drug discovery process, offering a cost-effective and time-efficient method of identifying potential drug candidates with high precision. It eliminates the need for time-consuming and resource-intensive experimental methods by providing a rational and systematic approach to selecting compounds with the greatest chances of success (Rao and Srinivas 2011, Shaker et al., 2021). This study focuses on computational screening of the drug library to identify potential PRKG1 inhibitors using a synergistic approach that combines virtual screening, molecular docking, and molecular dynamics (MD) simulation.
The crystal structure of the PRKG1 (PDB ID: 3OCP) was obtained from PDB and prepared. The residues of the PRKG1 protein's binding pocket were parameterized inside a three-dimensional grid box. Based on the binding affinities, a structure-based virtual screening of the pharmacological library of 150,000 compounds was conducted, and 100 compounds in total were evaluated against the PRKG1. The hit compounds identified during virtual screening were docked to the prepared PRKG1 receptor to predict binding affinities using the standard precision method of glide tool. This step aids in predicting the compounds' potential efficacy in inhibiting PRKG1 enzymatic activity(Jakhar et al., 2020). The top ten compounds were chosen for further investigation based on their binding affinities. The selected compounds had binding affinities ranging from −8.69 to −7.58 kcal/mol. The binding affinities of the selected compounds suggested that they could inhibit the function of the PRKG1 protein.
We examined the molecular interactions between the selected hits and the binding pocket of the PRKG1 receptor. Alkyl, Pi-Sigma, Pi-Pi Stacked, Pi-Sulfur, van der Waal, and conventional hydrogen bonds / carbon hydrogen bonds were the most often observed interactions. These interactions are critical in determining each of the top candidate compounds' binding affinities and docking scores.
Following the molecular interaction analysis, the top two compounds' plausible binding modes were investigated by aligning them on the co-crystal ligand. The docked compounds were perfectly aligned on the co-crystal ligand with similar binding mode, according to the analysis.
Using Molecular Dynamics (MD) simulations, the dynamic behavior of PRKG1 inhibitor complexes over time is also examined. This computational method can be used by researchers to examine the flexibility and stability of binding interactions, which offers important insights into structural alterations that could impact the effectiveness of the inhibitor (Salo-Ahen et al., 2020). Molecular dynamics simulations revealed that these compounds remained strong inhibitors within the protein binding region. All these findings suggest that the chosen hit compounds can function as lead compounds in inhibiting PRKG1′s biological activity.
5 Conclusion
In this comprehensive in silico study, we used MTI OPEN screening, molecular docking, and MD simulations to identify potential PRKG1 inhibitors successfully. The combination of these computational methods provides a solid foundation for the rational design of novel therapeutic agents targeting PRKG1. Further experimental validation of the identified compounds is required to proceed with these promising candidates further into clinical trials.
CRediT authorship contribution statement
Abdullah R. Alanzi: Writing – review & editing, Validation, Supervision, Conceptualization. Bayan Abdullah Alhaidhal: Writing – original draft, Formal analysis, Data curation. Fatimah Mohammed Alsulais: Writing – original draft, Software, Methodology.
Acknowledgement
Authors extend their appreciation to researchers supporting project Number (RSPD2024R885) at King Saud University Riyadh Saudi Arabia for funding this research.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Bowers, K. J., E. Chow, H. Xu, et al., 2006. Scalable algorithms for molecular dynamics simulations on commodity clusters. Proceedings of the 2006 ACM/IEEE Conference on Supercomputing.
- cGMP-dependent protein kinases as potential targets for colon cancer prevention and treatment. Future Med. Chem.. 2010;2(1):65-80.
- [Google Scholar]
- Thermodynamics and Kinetics of Drug-Target Binding by Molecular Simulation.. 2020;120(23):12788-12833.
- Mechanistic Insights into the Japanese Encephalitis Virus RNA Dependent RNA Polymerase Protein Inhibition by Bioflavonoids from Azadirachta Indica.. 2021;11(1):1-13.
- PRKG 1 and genetic diagnosis of early-onset thoracic aortic disease. Eur. J. Clin. Invest.. 2016;46(9):787-794.
- [Google Scholar]
- Comparison of MM/GBSA Calculations Based on Explicit and Implicit Solvent Simulations.. 2013;15(20):7731-7739.
- Henning, P., 2022. Functional characterization of disease-associated mutations in type I cGMP-dependent protein kinase.
- Relevance of molecular docking studies in drug designing. Curr. Bioinform.. 2020;15(4):270-278.
- [Google Scholar]
- The Role of Exosome and Protein Kinase cGMP-Dependent 1 (PRKG1) in Detecting of Breast Cancer in Patients with Breast Tumors. J Biomed Sci.. 2023;12(1):94.
- [Google Scholar]
- Kumar, S. J. J. o. B. and T. s. World, 2019. Comparative Modeling, Virtual Screening and Docking of CCR5 with Diverse Libraries: An Initiation towards Rational Drug Design to Identify a New Chemical Entity to Treat HIV. 8 (7) 1-4.
- Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments.. 2013;27(3):221-234.
- Martínez, L. J. P. o., 2015. Automatic identification of mobile and rigid substructures in molecular dynamics simulations and fractional structural fluctuation analysis. 10 (3) e0119264.
- Discovery of Fungal Denitrification Inhibitors by Targeting Copper Nitrite Reductase from Fusarium Oxysporum.. 2017;57(2):203-213.
- E.L. Norton D. Gordon B. Yang Managing the aorta in patients with a PRKG1 mutation J. Thorac. Cardiovasc. Surg. 157 4 2019 e107 e9.
- Pacheco, A. B. and L. J. B. R. L. S. U. Hpc, 2012. Introduction to AutoDock and AutoDock tools.
- Price, D. J. and C. L. J. T. J. o. c. p. Brooks III, 2004. A modified TIP3P water potential for simulation with Ewald summation. 121 (20) 10096-10103.
- Modern drug discovery process: An in silico approach. Journal of Bioinformatics and Sequence Analysis.. 2011;2(5):89-94.
- [Google Scholar]
- Sadeer, N. B., E. J. Llorent-Martínez, K. Bene, et al., 2019. Chemical profiling, antioxidant, enzyme inhibitory and molecular modelling studies on the leaves and stem bark extracts of three African medicinal plants. 174 19-33.
- Molecular dynamics simulations in drug discovery and pharmaceutical development. Processes.. 2020;9(1):71.
- [Google Scholar]
- Sargsyan, K., C. Grauffel, C. J. J. o. c. t. Lim, et al., 2017. How molecular size impacts RMSD applications in molecular dynamics simulations. 13 (4) 1518-1524.
- Protein kinase G1 regulates bone regeneration and rescues diabetic fracture healing. JCI Insight.. 2020;5(9)
- [Google Scholar]
- In silico methods and tools for drug discovery. Comput. Biol. Med.. 2021;137:104851
- [Google Scholar]
- Improving the Prediction of Absolute Solvation Free Energies Using the next Generation OPLS Force Field.. 2012;8(8):2553-2558.
- Thillainayagam, M., K. Malathi, S. J. J. o. B. S. Ramaiah, et al., 2018. In-Silico molecular docking and simulation studies on novel chalcone and flavone hybrid derivatives with 1, 2, 3-triazole linkage as vital inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. 36 (15) 3993-4009.
- Trott, O. and A. J. J. J. o. c. c. Olson, 2010. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. 31 (2) 455-461.




