

Translate this page into:
Characterization of the glyceraldehyde-3-phosphate dehydrogenase gene from the desert plant Haloxylon salicornicum using RT-PCR amplification and sequencing
⁎Corresponding author. s.almazrooei@ku.edu.kw (Suad S. Al Mazrooei)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Reverse transcriptase polymerase chain reaction (RT-PCR) is the most common molecular downstream application assay used to assess both quality and quantity of mRNA transcript, which heavily relies on using a sequence of an endogenous gene as a reference. In organisms with unknown genome sequence, designing of mRNA-specific PCR primers based on available sequence information of other species is an extremely useful approach to isolate homologous/orthologous genes. Here we report the isolation of a partial coding sequence of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene ortholog in Haloxylon salicornicum, a desert plant native to Kuwait, by designing a non-degenerate consensus primer pair based on conserved regions generated by a multiple alignment of orthologous GAPDH DNA sequences derived from closely-related species. Using both total RNA and genomic DNA extracted from leaf-stem segments, we proved that the designed primer pair is cDNA/mRNA specific, and there was no evidence of non-specific signals. Sequence and bioinformatics analyses of the obtained DNA fragment and its corresponding deduced amino acid sequence strongly suggested that it is a member of the cytosolic GAPDH family and that it is highly conserved. Based on the DNA sequence of the above-mentioned fragment, we also describe the RT-PCR amplification of a small-sized amplicon using a primer-template mismatch at the 3′-end region. The amplified products will be useful for PCR-based assays and gene expression studies of H. salicornicum, and it will be of great value to geneticists as well as evolutionary biologists.
Keywords
Haloxylon salicornicum
GAPDH
RT-PCR
Consensus primers

1 Introduction
Gene expression studies employ various molecular downstream applications, the most common of which is the reverse transcriptase polymerase chain reaction (RT-PCR) assay. The reliability of such assay depends on the availability of an endogenous reference sequence(s), which is often used either as a control for checking the integrity of the reverse-transcribed RNA sample (qualitative) or as a housekeeping gene for studying the regulated expression of other target genes (quantitative). The nuclear gene-encoded glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is frequently used as a reference (control) gene in both qualitative and quantitative RT-PCR assays. In higher plants, two classes of GAPDH protein are known that both show low sequence similarity to each other, a chloroplast-derived enzyme of the Calvin cycle (GAPA and GAPB; EC 1.2.1.13) and a cytosolic-derived glycolytic enzyme (GAPC; EC 1.2.1.12) (Cerff and Chambers, 1979). The glycolytic (catabolic) GAPC enzyme, in particular, is a tetrameric NAD-binding enzyme involved in glycolysis and glyconeogenic. It consists of two domains, a binding-terminal domain (Gp_dh_N), which is a Rossmann NAP(P) binding fold, and a C-terminal domain (Gp_dh_C), which is a mixed alpha/antiparallel beta fold (Seidler, 2003).
Genes intended for use as controls in RT-PCR assays are assumed to be informative and also easily directed during various amplification steps (Lion, 2001). This is a pivotal point in gene expression analysis to obtain reliable results. Several studies on GAPDH raised concerns about its use as an internal reference in gene expression studies due to the unstable expression of GAPDH gene under a variety of conditions (Oliveira et al., 1999; Thellin et al., 1999; Schmittgen and Zakrajsek, 2000; Suzuki et al., 2000). However, most of these studies involved GAPDH genes from organisms other than plants (i.e. human and mouse tissues). In plant-related gene expression studies, GAPDH is still used as internal control, probably due to several reasons. The most important of which is that GAPDH orthologous proteins are highly conserved across a wide range of plant taxa (Martin and Cerff, 1986). In addition, expression of GAPDH gene under various environmental (abiotic) stresses was found constitutive, stable, and occurs in different tissues and growth stages of plants (Iskandar et al., 2004; Hong et al., 2008; Bao et al., 2016). However, poor stability of this gene in plants has been reported (Castonguay et al., 2015; Zhang et al., 2017).
A common problem associated with the use of GAPDH as a reference gene is the presence of processed pseudogenes (Hurteau and Spivack, 2002; Liu et al., 2009; Sun et al., 2012). Such a problem is considered a critical issue in RT-PCR amplification of a particular gene due to the possibility of generating false positive results (Garbay et al., 1996). Nevertheless, this problem can be overcome by designing PCR primers in such a manner as to permit differentiation between cDNA- and genomic (gDNA)-derived PCR products, which in turns requires the availability of genomic sequence information to determine the appropriate location of the primers. Otherwise designing suitable primers would be a challenging task.
In organisms with unknown or limited genome sequence information, isolating homologous sequences of a particular gene family member is often achieved by designing degenerate consensus primers derived from reverse-translated sequences of highly conserved amino acids regions. Such regions are generated by performing multiple amino acids alignments or assembly of protein blocks that are based primarily on known sequences of distantly-related species (Pietrokovski, 1996; Rose et al., 1998). In contrast, designing non-degenerate consensus primers derived by choosing the most common nucleotide at every position across highly conserved regions is a more convenient approach to isolate unknown sequence based on multiple nucleotide alignments of functionally-related proteins (Smith et al., 1990). Compared to degenerate primers, non-degenerate consensus primers usually require fewer optimization steps, and it is more likely that the isolated gene product corresponds to the gene of interest.
We report the RT-PCR amplification and isolation of GAPDH homologous/orthologues partial sequence using total RNA extracted from leaf-stem segments of the Kuwaiti native desert plant Haloxylon salicornicum (Moq.) Bunge ex Bioss. (Amaranthaceae) using a consensus (non-degenerate) primer pair that was designed based on known orthologous GAPDH coding DNA sequences of closely-related species. Our approach combines and modifies some of the above-mentioned strategies. Several appropriate controls to validate the specificity of designed primers for amplification of targeted cDNA were employed, and its identity was confirmed by sequencing. We also describe the RT-PCR amplification of a small-sized amplicon using primer-template with and without mismatch based on the sequence information of the obtained GAPDH partial sequence mentioned above.
2 Materials and methods
2.1 Primer design
Primer designing strategy was based on the alignment of putative orthologous GAPDH sequences across multiple species that share similar functional and structural properties. Screening for putative homologous GAPDH genes available in the databases was performed according to the two following criteria: i) only cytosolic-origin mRNA coding sequences (cds) were selected, while those chloroplast-origin mRNA sequences or DNA-derived sequences were excluded; ii) when searching was done against RefSeq mRNAs, we went in the favor of curate genebank entries, whereas model mRNA sequences that resulted from computational prediction of genome annotation were not considered. Accordingly, we searched first for GAPDH mRNA sequences from species of the family Amaranthaceae. However, only two cytosolic GAPDH mRNA complete cds were available in the GeneBank database: (1) from Atriplex nummularia (Accession No. U02886; Old Man Saltbush hereafter) and (2) from Beta vulgaris subsp. vulgaris (Accession No. EF408234; Red Beet hereafter). These sequences were retrieved from NCBI database and exported into our licensed CLC Workbench 8.0 software (Katrinebjerg, Aarthus, Denmark) as FASTA files. Nucleotide sequences were translated into amino acids using the CLC software. Pairwise comparison and searching in conserved domain database (CDD) (Marchler-Bauer et al., 2007) proved that the retrieved GAPDH sequences belong to the cytosolic GAPDH protein family (PROSITE PS00071; EC. 1.2.1.12). Initially, we used BlastX to search for homologous proteins in both non-redundant (nr) and UniProt/SwissProt databases at NCBI using the coding sequence of Red Beet as the main query. To infer that two proteins are homologous, an E-value cutoff of 1e-3 was used. Since we are interested in detecting closely-related members of the GAPDH protein family rather than finding all potential similarities, we chose BLOSUM 80 scoring matrices (Henikoff and Henikoff, 1992) with gap cost 11/1; however using BLOSUM 62 did not reveal significant differences. The search resulted in a relatively large number of homologous proteins with statistically significant sequence similarity (E-value < 10e-3; bit score > 50) (data not shown). Performing the same search using the coding sequence of Old Man Saltbush resulted in almost similar hits. Domain structure was examined by performing Pfam domain search (http://pfam.xfam.org) with an E-value of 0.001 to identify any possible unrelated sequences. Eventually, inferring functional similarity based on conserved domains and signature patterns revealed several protein-coding sequences that share similar enzymatic function (EC 1.2.1.12), and contained the GAPDH protein family pattern [ASV]-S-C-[NT]-T-{S}-x-[LIM] (PS00071) where cysteine (C) is the active site (Fabry et al., 1989) (data not shown). The selected ortho–homologs proteins were back translated into nucleotide sequences, and then globally aligned using ClustalW (Thompson et al., 1994) to identify consensus regions (Supplementary Fig. S1). Regions that share 100% identical DNA sequences were observed only between GAPDH protein sequences from Amaranthaceae species. This was not the case when other closely-related species were included, from which sequence-specific primers could be designed to amplify via PCR the ortholog GAPDH sequence from Red Beet and Old Man Saltbush, and would be more likely to amplify the same ortholog in H. salicornicum. Accordingly, conserved regions I (36 bases), II (20 bases) and III (38 bases) that serve as a target for PCR primer designing were selected (Fig. 1). A group of candidate primers were first checked for any possible internal repeat matching, primer-dimer or harpin formation, and their melting temperatures were calculated using the CLC software. Best primer pairs showed acceptable parameters were chosen (Table 1). Specificity of each primer was also tested by Primer Blast against ‘nr’ database available at the NCBI website (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). The designed primers did not reveal matches with sequences different from cytosolic GAPDH mRNA. Primer specificity was further checked using the thermodynamics-based MFEprimer-2.0 program (Qu et al., 2012). The three primers S1, S2 and S3 have an identical match (sequence-specific) on the GAPDH mRNA coding sequences from the Amaranthaceae species only, while they have an incomplete match (consensus) on the GAPDH mRNA coding sequences derived from closely-related species belong to different families. We refer to such primer sets as sequence-specific primers for the family Amaranthaceae. The primer pair S1/S2 was designed to amplify and clone the mid-fragment of the GAPDH gene from H. salicornicum covering the area between regions I and II, whereas the primer pair S1/S3 was designed to amplify larger fragment covering the area between regions I and III. Based on GAPDH mRNA complete cds from the two closely-related species, Old Man Saltbush and Red Beet, the designed primer sets S1/S2 and S1/S3 would potentially amplify fragments with a size of 171 bp and 460 bp, respectively. In addition, a gene-specific primer S4 (Table 1) was designed based on the sequence of the GAPDH fragment obtained by PCR amplification using the primer pair S1/S3 (shown later). The designed oligonucleotide primers were synthesized commercially at Biomers GmbH (Ulm, Germany).
Regions I, II and III selected from the alignment of back-translated DNA sequences of homologous orthologs GAPDH protein coding regions (see supplementary file S1) which served as a target for PCR primer designing. An arrow indicates the position and direction of the designed primers. The numbers denote the start and end position of the primers relative to Red Beet GAPDH coding region. Beta vulgaris subsp. vulgaris (Acc. No. EF408234); Atriplex nummularia (Acc. No. U02886); Mesembryanthemum crystallinum (Acc. No. J05223); Arabidopsis thaliana (Acc. No. M64116), Petroselinum crispum (Acc. No. X60344), Ranunculus acris (Acc. No. X60345), Antirrhinum majus (Acc. No. X59517), Pisum sativum (L07500); Glycine max (Acc. No. NM_001249200), Cucumis sativus (Acc. No. NM_001280743), Solanum lycopersicum (Acc. No. NM_001247874), Magnolia liliiflora (Acc. No. X60347), Taxus baccata (Acc. No. L26922), Ginko biloba (Acc. No. L26924) and Pinus sylvestris (Acc. No. L07501).
Primer
Length (nt)
Direction
Sequence (5′ → 3′)*
S1
20
Forward
GTACGACAGTGTTCACGGTC
S2
20
Reverse
GTGAAGACACCAGTGGACTC
S3
20
Reverse
TCCACCTCTCCAGTCCTTAG
S4
20
Reverse
GTGAAGACACCAGTGGATTC
2.2 Total RNA extraction and quality control
Total RNA was extracted from H. salicornicum leaf-stem tissues using the RNeasy Plant Mini Kit (Qiagen, Valencia, CA), followed by RQ 1 RNase-free DNase I (Promega, Madison, WI) to eliminate any contaminating gDNA. Total RNA was then cleaned up from the enzymatic reaction and concentrated by silica-based spin columns using RNeasy MiniElute Cleanup Kit (Qiagen) following the manufacturer’s instructions. The amount of both isolated total RNA and residual gDNA that remained in the RNA preparation after DNase treatment was measured by Qubit 2.0 Fluorometer using the RNA BR Assay Kit (20–1000 ng) and the dsDNA HS Assay Kit (0.2–100 ng), respectively (Life Technologies, Carlsbad, CA, USA). Purity of the RNA samples was checked by electrophoresis analysis using non-denaturing 1.2% agarose gel stained with ethidium bromide (EtBr) in 1X TAE (Tris-acetate EDTA) running buffer, and then visualized under UV illumination. Gel images were captured by the Image Lab software bundled with the Gel Doc EZ Gel Imaging System (BioRad, Hercules, CA, USA). Moreover, RNA integrity number (RIN) was determined using the Agilent RNA 6000 Pico Chip Kit with the Agilent 2100 Bioanalyzer (Agilent Biotechnologies, Santa Clara, CA, USA). For cDNA synthesis, only RNA samples that showed ribosomal RNA ratio (28S/18S) of ≥1.8, and RIN of ≥8.0 with low gDNA content were selected.
2.3 RT-PCR, cloning and sequencing of a partial cDNA fragment of GAPDH gene
The primary cDNA fragment of the GAPDH gene (covering the area between region I and III) was synthesized simultaneously following two different approaches. In the first method, poly A+ mRNA was isolated from total RNA with Oligotex mRNA Mini Kit (Qiagen). Then, approximately 2 µg of the purified mRNA was used for first- and second-strand syntheses with random hexameric primers in a total reaction volume of 25 µl using the Universal RiboClone cDNA Synthesis System (Promega) in accordance with the manufacturer’s protocol. In the second method, cDNA was synthesized from approximately 2 µg of total RNA with the reverse primer S3 in a total reaction volume of 20 µl using the GoScript Two Steps RT-PCR System followed by the RNase H step (Promega). A reaction tube containing total RNA extracted from Red Beet was included as a positive control, and reaction tubes without the addition of the template (NTC) or RT enzyme (RTC) were included as negative controls. cDNA covering the area between regions II and III was synthesized using sequence-specific reverse primers S2 and S4. PCR reaction was performed in a total volume of 50 µl, including 5 µl of 5X PCR buffer (containing 20 mM MgCl2), 2 µl of dNTP mix (25 mM each), 1 µl of forward primer (10 mM), 1 µl of reverse primer (10 mM), 3 µl of template cDNA, and 0.5 µl of GoTaq DNA polymerase (500 units, Promega). The PCR amplification was conducted on the 9700 Thermal Cycle (Life Technologies), and amplification conditions for primer pair S1/S3 were: initial hot start cycle at 95 °C for 5 min during which the polymerase was added, followed by 35 cycles consisting of denaturation at 94 °C for 30 s, primer annealing at 52 °C for 30 s, primer extension at 72 °C for 1 min, and final extended elongation cycle at 72 °C for 6 min. To rule out any non-specific amplification that may result from gDNA contamination in the RNA preparations, two additional reaction tubes were included as controls. The first tube contained column-purified total RNA which was pre-treated with RNase A (4 mg/ml; Promega) prior to subjecting to reverse transcriptase reaction, whereas the other reaction tube contained gDNA of the same source from which RNA was extracted and pre-treated with RNase A. Both additional control tubes were subjected in parallel to PCR amplification using same parameters that used with primary cDNA templates (described above). The same PCR conditions (described above) were used with primer pairs S1/S3 and S1/S4 except that the annealing temperature was reduced to 48 °C. Reproducibility of the selected primers was evaluated by performing at least three independent PCR reactions using newly synthesized cDNA each time, and all amplification products were size-separated by gel electrophoresis. PCR-amplified fragments were gel-purified using the QIAprep Gel Extraction Kit (Qiagen), and were T/A-cloned into pGEM-T-easy vector (Promega) following the manufacturer’s protocol. Recombinant clones were transformed to Escherichia coli JM109 competent cells (Promega), and streaked on ampicillin/Xgal/IPTG SOB medium plates. Positive recombinant clones were selected by blue-white color selection and were screened for the presence of inserted DNA of the expected length by EcoR I restriction enzyme digestion (Promega). Plasmids DNA were purified from bacteria cells by QIAprep Spin Miniprep kit (Qiagen), and were sequenced from both directions by the BigDye Terminator v3.1 sequencing kit (ThermoFisher Scientific, Waltham, MA, USA) using the ABI 1130xl Genetic Analyzer (ThermoFisher Scientific) available in the Biotechnology Center at Faculty of Science, Kuwait University. At least 10 positive clones of each PCR product were sequenced, and sequencing was repeated twice using newly synthesized cDNA.
2.4 Sequence analysis
Unless otherwise stated, sequence analyses were carried out using our licensed CLC Genomic Workbench. All individual sequences were first trimmed to exclude low quality sequences using a cutoff score limit of 0.001 as well as vector match sequences. Subsequently, forward and reverse DNA sequence strands of each individual clone were assembled to contig to obtain clone consensus sequence. One consensus sequence was finally generated by assembling consensus sequences of all clones and to record any possible conflicts. Obtained sequences were translated into protein (reading frame +2) using the standard genetic code. Searches for nucleotide and amino acid sequence similarities were conducted using the BLAST tool (Altchul et al., 1997). Obtained sequences were also compared against its original query using BLAST2Seqs (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The deduced amino acid sequence was aligned with the GAPDH gene coding regions from other species by ClustalW using the default parameters. Domain sequences in the deduced amino acid were predicted using the Pfam domain searching tool (as described above). The functional site of the deduced amino acid sequence was identified using the PROSITE program (http://www.expasy.org/prosite).
3 Results
3.1 RT-PCR amplification of a partial sequence of the GAPDH gene using consensus primers specific to the family Amaranthaceae
Results in Fig. 2 showed that the primer pair S1/S3 (targeting the sequence between regions I and III) produced a single fragment of about 400–500 bp when a cDNA template synthesized by the sequence-specific method was used. As expected, a 460 bp fragment of GAPDH was produced when total RNA extracted from Red Beet was used as a template (whose sequence was included in the alignment used for designing the primers). When random-primed cDNA was used as a template, the same fragment size was produced with almost equal yield and efficiency to that obtained with the sequence-specific primer cDNA. Negative control reactions (NTC and RTC) did not show any non-specific bands after 35 cycles in both cases. The total RNA purified by the MiniElute column contained very low gDNA residues (∼16 ng/µl) (data not shown). To exclude the possibility of gDNA interference with RT-PCR assay, an aliquot from the same total RNA that was used in the RT-PCR assay was treated with RNase A before subjecting to reverse transcriptase reaction. No amplification product was seen under the assay conditions employed, indicating that the remaining amount of gDNA in the RNA preparation did not interfere with RT-PCR amplification of the cDNA target. Furthermore, when gDNA extracted from the same source of that used for cDNA synthesis was treated with RNase A prior to subjecting to the same end-point conventional PCR conditions that were used with RNA template, no band was observed on the agarose gel. This result eliminates the possibility of false-positive signals that may derive from contaminating gDNA; e.g. co-amplification of processed pseudogenes.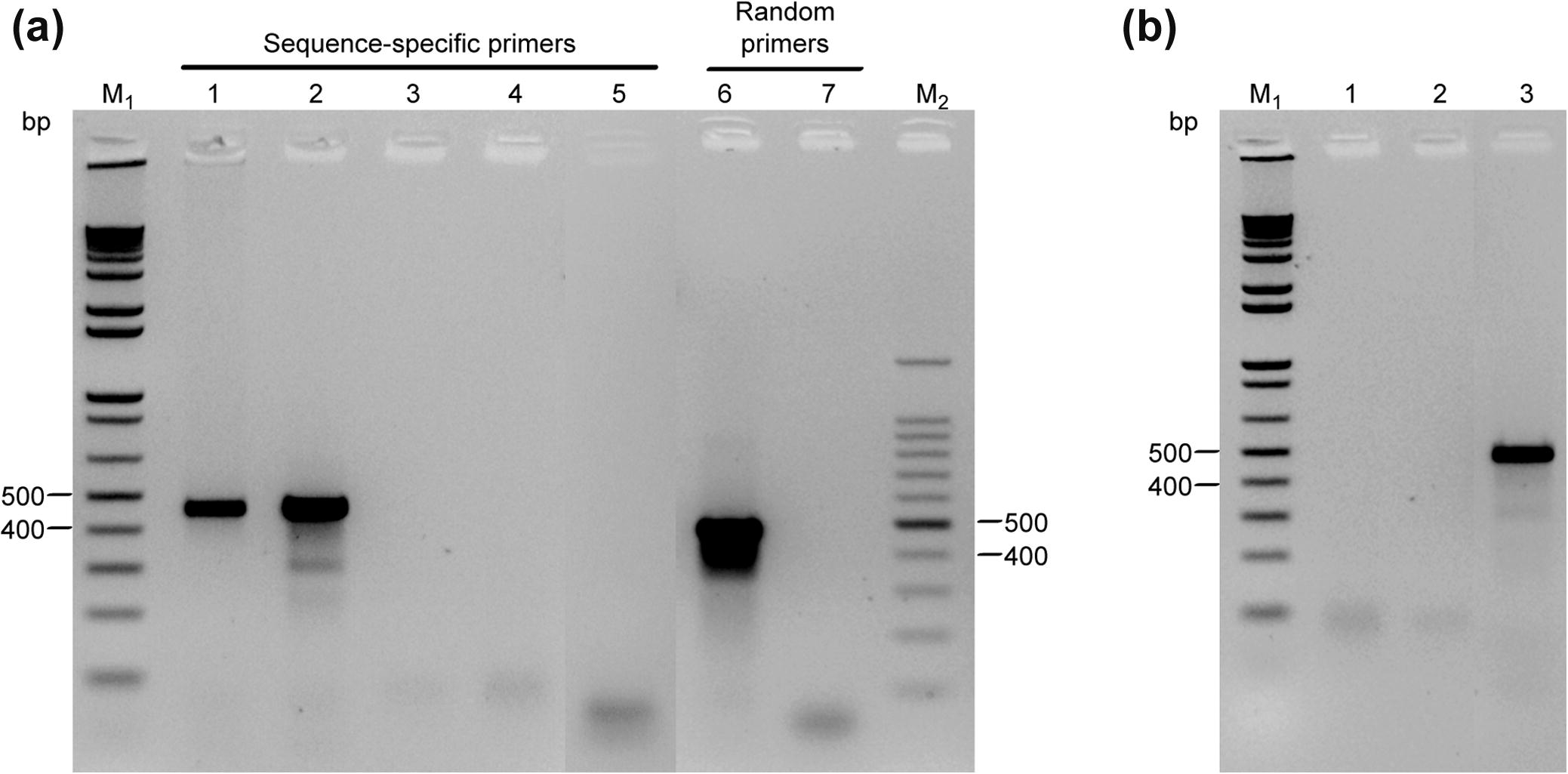
(a), RT-PCR amplification of a partial sequence of the cytosolic GAPDH gene. cDNAs were synthesized from total RNA extracted from leaf-stem tissues and primed with either sequence-specific primer pair S1/S3 or random hexamer primer. Lane 1: positive control B. vulgaris subsp. vulgaris; Lanes 2 and 6: H. salicornicum; Lane 3: no-template control (NTC); Lanes 4 and 7: reverse-transcriptase controls (RTC); Lane 5: total RNA from H. salicornicum pre-treated with RNase A (negative control); (b), End-point PCR amplification control of GAPDH gene from H. salicornicum with primer pair S1/S3. Lane 1: gDNA template extracted from the same source of that used for cDNA template synthesis; Lane 2; NTC; Lane 3: synthesized cDNA from total RNA used as a template (positive control). PCR products were analyzed by electrophoresis in 1.5% agarose gel and visualized by ethidium bromide staining. Each lane was loaded with 5 µl of the PCR reaction. M1: 1 kb DNA ladder (Promega); M2: 100 bp DNA ladder (ThermoFisher Scientific); bp: base pair.
To confirm the identity of the PCR products, the fragment was T/A cloned into pGEM-T Easy vector using the A-overhang produced by Taq DNA polymerase and subsequently sequenced. Sequence analysis of randomly selected clones revealed that the size of obtained partial gene sequence is 460 bp and that all sequenced clones were flanked with both S1 and S3 primers, which were identical to the designed ones. Based on the position of the primers relative to known sequences from Old Man Saltbush and Red Beet; this fragment is of the expected size. The cloned fragment was designed GAPDH460 (Fig. 3). Results of a BLASTN search showed that GAPDH460 shared high homologies with the cytosolic GAPDH gene from other species (data not shown). This strongly suggests that the GAPDH460 sequence cloned from H. salicornicum belonged to GAPDH family. The nucleotide sequence of the mRNA partial sequence of GAPDH cDNA obtained in this study was deposited to the GenBank database under Accession No. KU723650.
The nucleotide and deduced amino acid sequence of H. salicornicum GAPDH460. The forward S1 and reverse S3 primers sequences are shown (delimited by arrows). The characteristic signature pattern of GAPDH family is underlined. A conserved cysteine residue (C) that is important for the binding of glyceraldehyde-3-phosphate in the catalytic site is indicated with an asterisk (*).
The GAPDH460 partial sequence was found to code for 153 amino acid residues (Fig. 3). BLASTP and BLASTX results showed that the obtained deduced amino acid was highly homologous to those GAPDH genes that were used in constructing the alignment, particularly with GAPDH from the Amaranthaceae species Old Man Saltbush and Red Beet. This proves that the GAPDH fragment obtained is a member of the GAPDH family. Multiple sequence alignments of GAPDH460 amino acid sequence with known GAPDH from several plant species revealed that they were highly conserved, especially in the regions of the GAPDH family signature (Fig. 4). Comparison of the deduced amino acid of GAPDH460 with those from the Amaranthaceae family indicated that the sequence identity was 91.23% and 90.45% with Old Man Saltbush and Red Beet, respectively (Fig. 5). Only one non-synonymous substitution occurred in the GAPDH460 deduced amino acid, alanine (A), instead of threonine (T) and serine (S) in the Old Man Saltbush and Red Beet protein sequences, respectively. A conserved domain search against the CDD database at the NCBI of the partial deduced protein sequence showed the presence of two domains, the Gp_dh_N domain (pfam 00044; E-value 4.95e-50) and the Gp_dh_C domain (pfam02800; E-value 3.87e-22), and a conserved cysteine (C) active site (Fig. 3). Also, analysis of the deduced amino acid sequence by SMART resulted in the same above-predicted domains with high a significance score (data not shown). This high degree of similarity of both identified domains in GAPDH460 to the known domains of GAPDH protein suggests that they share the same function, which support our results that the obtained GAPDH partial sequence does indeed belong to the glycolysis GAPDH protein family.
ClustalW alignment of GAPDH460 deduced amino acid sequence from H. salicornicum against selected GAPDH sequences from other species. Gaps have been introduced to permit alignment. The graphic indicates to the degree of conservation between aligned sequences estimated by the tall of column. The red box indicates GAPDH family signature. Species with the GenBank accession numbers are indicated between parentheses.

Alignment of GAPDH460 deduced amino acid with corresponding regions in the protein coding sequences of the Amaranthaceae species Old Man Saltbush and Red Beet. Light gray shaded residues indicate synonymous substitutions. Dark gray shaded residue indicates the only nonsynonymous substitution. Proteins accession numbers AAA03442 is Atriplex nummularia (Old Man Saltbush), ABN50381 is Beta vulgaris subsp. vulgaris (Red Beet), and AML22888 is the deduced amino acid from Haloxylon salicornicum obtained in this study.
3.2 RT-PCR amplification of a GAPDH amplicon using primer-template mismatch
The obtained sequence of the GAPDH460 clone revealed that the reverse primer S2 contains a single T/C mismatch near its terminal 3′ base (G/A on the sense strand of the GAPDH460 DNA sequence). To study the effect of this primer-template mismatch on RT-PCR efficiency, we attempted to amplify a 171 bp DNA fragment of GAPDH from H. salicornicum using the normal upstream primer S1 in combination with the downstream primer S2 (corresponding to bases 1–171 in the GAPDH460 sequence). In parallel, we used primer S1 in combination with another downstream primer (S4), which completely matched the GAPDH460 DNA template. Both primer pairs were used in the RT-PCR assay (as performed for GAPDH460 amplification) with low annealing temperature (48 °C). Using the normal primer pair S1/S4, the desired 171 bp DNA fragment was amplified satisfactory without any detectable non-specific products. Interestingly, the primer pair with mismatch (S1/S2) produced a similar result (Fig. 6). Sequence analyses revealed that both fragments do indeed correspond to the first 171 bases of the GAPDH460 fragment. However, assembly of sequences derived from at least 10 randomly selected clones revealed that approximately half the number of the clones obtained with the primer pair S1/S2 (containing the single mismatch) maintained the mismatched base, whereas the remaining number were 100% identical to the GAPDH460 sequence. As expected, primer pair S1/S4 (completely matched the DNA template) formed 20 normal base pairs with the template with no mismatch in all sequenced clones (data not shown).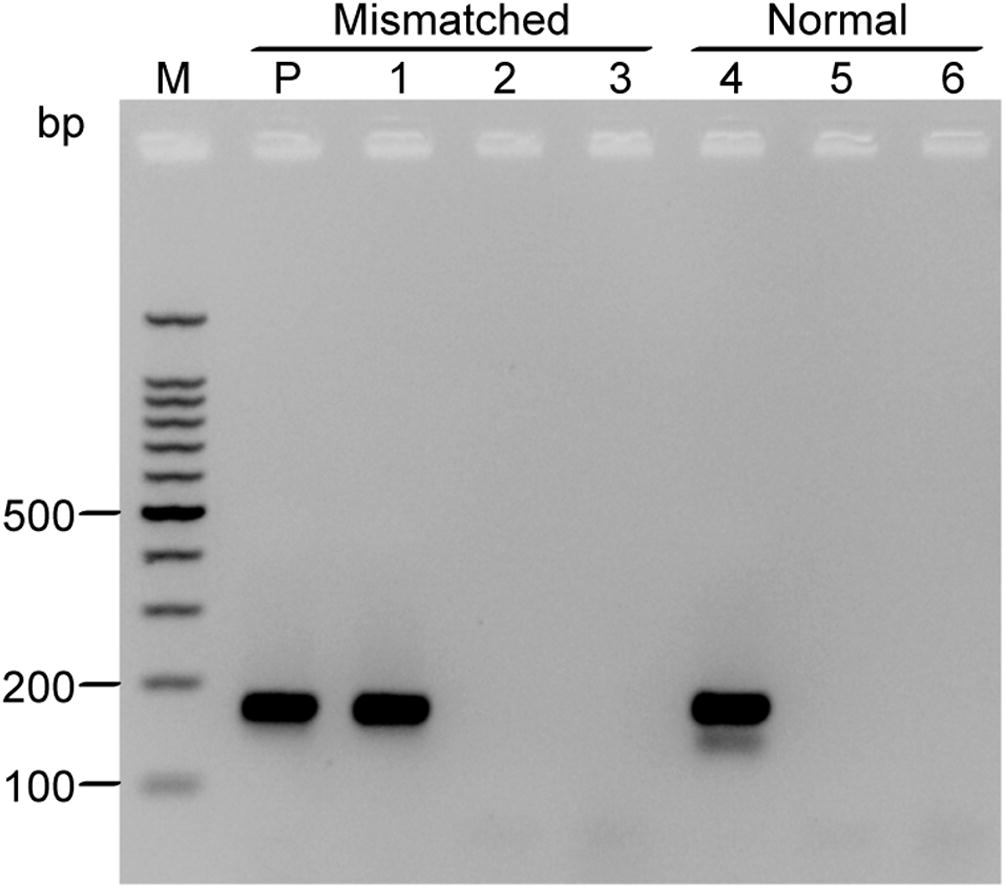
RT-PCR amplification of GAPDH171 fragment from H. salicornicum using total RNA extracted from leaf-stem tissues. cDNA was primed with either the primer pair S1/S2 that contains single mismatch near the 3′ region or with the normal primer pair S1/S4 that contains no mismatch. Total RNA extracted from leaf tissues of Red Beet was included as positive control (P). Lanes 1 and 4 are total RNA from H. salicornicum treated with DNase I prior subjecting to reverse transcriptase reaction. Lanes 2 and 5 are non-template controls (NTC). Lanes 3 and 6 are reverse transcriptase controls (RTC). Both primer pairs resulted in an amplicon with a size of 171 bp. PCR products were analyzed by electrophoresis in 2% agarose gel and visualized by ethidium bromide staining. Each lane was loaded with 5 µl of the PCR reaction. M: 100 bp DNA ladder (ThermoFisher Scientific); bp: base pair.
4 Discussion
Our main aim was to isolate the GAPDH orthologous gene from the Amaranthaceae species H. salicornicum for later use as a reference in the various molecular downstream applications related to our current as well as future studies; i.e. gene expression. Special emphasis was given to this plant as a representative of many native plants growing in the desert of Kuwait with unknown genomic information. Thus, we attempted to design oligonucleotide primers capable of specifically amplifying GAPDH partial sequences with desired lengths suitable for both qualitative and quantitative RT-PCR assays, rather than amplifying the full-length sequence. The latter is often required by studies interested in the evolution of a particular gene family. In organisms with limited genomic sequence information, it is often preferable to use nested primers to ensure that the correct gene family member is amplified. This strategy, however, would be necessary in the case of degenerate PCR. Fredslund et al. (2005) developed a program useful for PCR amplification of orthologous sequences in species where prior sequence information is not available based on alignment of DNA sequences from phylogenetically related species. However, this program was designed to find primers in specialized alignments where at least one of the sequences has annotated introns, and the output is a list of degenerate primers. In the present work, we have employed a PCR primer design strategy that is based on nucleotide sequences of closely-related species to design consensus non-degenerate primers, rather than degenerate ones. However, there is a very small chance to find consensus sequence-specific primers across multiple species for a certain gene-family member, and this is heavily reliant on sequences available from other related species and the type of target gene. Therefore, we first focused on finding homologous orthologs of the cytosolic GAPDH mRNA sequences from closely-related species, including those belonging to the family Amaranthaceae. Our interest was to look for those primers that are conserved across multiple species phylogenetically close to the species under study and that are also specific for amplifying the cytosolic-derived glycolytic enzyme class (GAPC; 1.2.1.12). We searched for patterns of conserved amino acid residues in groups of functionally related proteins, and we used BLAST similarity searching method to identify homologous proteins that share statistically significant sequence similarity. Orthologous proteins sharing the same function were identified using the CDD and Pfam searching tools to find conserved domains and, if possible, also structures. Information from both amino acid and nucleic acid sequences was used to produce the alignments. In the former, orthologous homologous GAPDH proteins were identified, whereas the latter was used to select possible candidate primers. We are confident that the alignments used in this study are unlikely to contain unrelated sequences since the statistical estimate of the BLAST analysis rarely obtains expectation values lower than ones expected by chance (Pearson, 1995). Moreover, since the DNA sequences used in the alignment were intronless, the likelihood that the high similarity obtained in the alignments extends into a non–homologous intron or flanking sequences is negligible. This will ensure that there is no intron within the primer sites, and thus increasing the success of the PCR amplification.
For ideal designing of a specific mRNA/cDNA primer pair that does not recognize pseudogenes in RT-PCR amplification, information about the genomic sequence of the species under study is necessary. Such information can be helpful; e.g. to distinguish duplicated pseudogenes from cDNA by size, but still not sufficient to distinguish intronless processed pseudogenes. Alternatively, a set of appropriate negative controls is commonly included along with the experimental samples. As the genomic sequence of H. salicornicum is not yet available, we were compelled to design consensus primers to amplify the GAPDH ortholog gene based on related GAPDH sequences from other closely-related species. Following the strategy described above, we have successfully amplified a short DNA sequence of the GAPDH gene using total RNA extracted from leaf-stem tissues of H. salicornicum by RT-PCR assay. The sequence obtained in this study is a partial sequence of the GAPDH gene located in the coding region of the protein, and its size was dependent on the location of the selected consensus regions within the coding region of the protein. Under the conditions employed in the present work, there was also no need for PCR optimization steps with the designed consensus primers, which is often the case when degenerated primers are used.
The results of the Qubit dsDNA high sensitivity assay proved that the complete removal of contaminating gDNA from RNA preparations could not be achieved by DNase treatment. This is in agreement with several previous reports (Hurteau and Spivack, 2002; Harper et al., 2003; Carraro et al., 2005). Cleanup of extracted total RNA through a spin column without DNase treatment was equally efficient to remove gDNA contaminants with less loss of RNA compared to DNase treatment (data not shown). In the present study, we have shown that the designed consensus primer pair S1/S3 is an mRNA/cDNA specific by using a set of appropriate negative controls. Both mRNA-specific and random primers resulted in a single specific band after RT-PCR amplification. The absence of any amplified signal from the gDNA template pre-treated with RNase A enzyme indicated that the primers might be either flanking a large-sized intron or one primer at least spans an exon/exon border. This conclusion is supported by the fact that no signal was observed in either the RTC sample or in the sample that contained total RNA pre-treated with RNase A enzyme, where traces of gDNA are supposed to exist. Furthermore, sequencing of the cloned cDNA fragment (GAPDH460) confirmed the identity of the amplified product as one of the GAPDH family sequences. Only one clone among more than 20 recombinant clones sequenced contained a single mismatch, probably due to an error in the sequencing process. The results, therefore, do not indicate the presence of multiple GAPDH genes in the plant genome (data not shown). All results shown above confirm that the consensus primer pair S1/S3 was capable of supporting amplification from GAPDH cDNA and not from gDNA-derived templates.
The results of the present study showed that the amplified GAPDH460 region is highly conserved, especially in GAPDH orthologs from the family Amaranthaceae. According to the GAPDH mRNA complete coding sequence from Old Man Saltbush and Red Beet, the primer pair S1/S3 would amplify a product with a size of 460 bp from both sequences. In our study, sequencing of the GAPDH fragment amplified from H. salicornicum using the same primer pair revealed that the product size was exactly 460 bp. The lack of any deletions or insertions in this region from the above sequences indicates that this region is highly conserved. The GAPDH460 region contained only one non-synonymous substitution, which supports the hypothesis that evolutionary changes in the nucleotide sequence are more frequent between protein members from different families than members of the same plant family (Brown, 2002). Analysis of conserved domains showed that GAPDH460 contained a highly conserved domain for NAD+ binding and catalytic function and also the characteristic protein family pattern. This indicates that the GAPDH gene in H. salicornicum is likely to be highly conserved in terms of function and structure.
Comparison of the GAPDH DNA sequence with other available sequences from members of the Amaranthaceae revealed that the conserved regions I and III (used to design primers S1 and S3 respectively) are identical and have not been subjected to any changes (at least for sequences corresponding to the designed primers). This indicates that these two regions might be important to a particular function carried by the exon. These identical nucleotide regions could be generated only when GAPDH protein-coding sequences from available Amaranthaceae species were aligned, whereas including GAPDH sequences from other closely-related species in the alignment did not reveal identical consensus regions. In contrast, region II was shown to be less conservative and has one synonymous mutation (A instead of G, base 154). The alignment showed that the sequence of the designed consensus primer pair S1/S3 is specific to cytosolic GAPDH genes from the Amaranthaceae species Old Man Saltbush and Red Beet. Interestingly, sequence analysis of GAPDH460 revealed that the sequence of primer pair S1/S3 is also specific to GAPDH from H. salicornicum. The successful amplification of same the sized-product of GAPDH460 from Red Beet using the same primer pair (S1/S3) suggests that the described primer pair might be specific to the family Amaranthaceae, and may work in a wide range of different species as well. Such a suggestion is supported by the fact that the GAPDH gene in plants is highly conserved and that GAPDH homologous sequences from the family Amaranthaceae share >80% identity. A study has reported on the successful amplification of a partial sequence of the vacuolar anion transporter CLCc gene ortholog in Lepidium sativum by designing gene-specific primers using the Arabidopsis thaliana mRNA nucleotide sequence information, a closely-related species belonging to the Brassicaceae (Rani et al., 2013). However, our analysis was based on only a few sequences from Amaranthaceae species that were available in the database. Therefore, further empirical studies are necessary to verify this assumption (family-specific primer pair) by trying to amplify GAPDH genes from more Amaranthaceae species using the same primer pair, and confirming their identity by sequencing.
The availability of a GAPDH partial sequence from H. salicornicum (GAPDH460) has allowed us to study the effect of primer-template mismatch on the efficiency of RT-PCR amplification. The effect of primer-template mismatches on RT-PCR efficiency has been explained in detail in several reports (Creighton et al., 1992; Christopherson et al., 1997). The degree of primer-template mismatch impact on RT-PCR efficiency is determined by several factors, mainly the type and position of the mismatch base in the primer. For example, mismatches located at the 3′ region (last 5 nucleotides) of the primer have significantly larger effects on priming efficiency more than 5′ located mismatches (Ayyadevara et al., 2000; Wu et al., 2009). Purine-pyrimidine mismatches have relatively minor effects on RT-PCR efficiency and are more stable compared to purine-purine mismatches (Stadhouders et al., 2010). On the other hand, Huang and Cloutier (2007) found that the reduction in PCR yield is correlated with the number of primer-template mismatches. In the present study, we used a pair of primers (20 nt in length each) that were either completely complementary to the GAPDH460 template (S1/S4), or had a single mismatch near the 3′ region of the reverse primer (S1/S2). Our results showed that the T/C mismatch primed the synthesis of a 171 bp specific PCR product almost as efficiently as the normal primer, as the PCR product yield of both amplifications was markedly high. Similar observations were reported by Stadhouders et al. (2010) who concluded that the effect of primer-template mismatch took place only during reverse transcription, and thus there was no effect on the subsequent PCR cycles. Since the mismatch exists in the reverse primer S2, we assumed that the generated cDNA would probably be identical to the sequence of primer S2. Interestingly, sequence results of GAPDH171 clones amplified using the primer pair S1/S2 revealed that about 50% of the clones were identical to the mismatched reverse primer (containing C:T), whereas the remaining clones completely matched the sequence of GAPDH460. This is probably attributed to the efficiency of M-MLV reverse transcriptase enzyme to extend mismatches. The use of a low annealing temperature (Tm < 50 °C) might also be one of the reasons for such tolerance. When the normal reverse primer S4 replaced the reverse primer S2, all obtained clones were identical to the GAPDH460 sequence and contained the correct base A. In cases where primer-template mismatches are unavoidable, this approach can still be useful in conventional RT-PCR but not in quantitative RT-PCR. Many reports have indicated the effect of primer-template mismatches on the accuracy of quantitative PCR (Whiley and Sloots, 2005; Bru et al., 2008; Ledeker and De Long, 2013). Therefore, further analysis is required to address this question using GAPDH171 obtained with or without primer-template mismatch. Unfortunately, we were not able to study the effect of primer-template mismatch on PCR yield because no amplification occurred when gDNA was used as a template.
In conclusion, this was the first report of RT-PCR amplification, cloning and sequence analysis of GAPDH partial sequences from H. salicornicum using mRNA/cDNA specific primers designed based on consensus regions within multiple aligned GAPDH coding DNA sequences from closely-related species. We proved that the amplified GAPDH reverse-transcribed mRNA was specific and not hampered by the pseudogene co-amplification problem. The size of the obtained GAPDH460 sequence was sufficient to enable its assigning to the GAPDH gene family using several bioinformatics approaches. The reported partial sequence of the GAPDH gene isolated from H. salicornicum will be useful in molecular downstream application studies of native desert plants in Kuwait in the near future. The 460 bp fragment amplified using the designed primer pair S1/S3 (GAPDH460) was optimized only for conventional RT-PCR, whereas the 171 bp fragment amplified using primer pair S1/S4 (GAPDH171) would be suitable for qRT-PCR and/or hybridization probes assays. However, the successful amplification of the GAPDH171 provides indirect evidence on the integrity of the sequence itself. Therefore, more experiments are needed to test the expression stability of GAPDH171, and validate its use as a reference gene in qRT-PCR assay under various environmental stress conditions.
Declaration of interest
The authors report no conflicts of interest.
Acknowledgments
This work was supported by the Research Grant No. (SL05/12), Research Sector (RS), Kuwait University (KU), Kuwait. We acknowledge the General Facility Project (GS01/02), Faculty of Science, KU, for sequencing services. We also acknowledge the Research Sector Projects Units (RSPU) (GS02/11) and the Biotechnology Center (BTC), both at Faculty of Science, KU, for DNA/RNA quality control analyses.
References
- Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res.. 1997;25:3389-3402.
- [Google Scholar]
- Discrimination of primer 3′-nucleotide mismatch by taq DNA polymerase during polymerase chain reaction. Anal. Biochem.. 2000;15:11-18.
- [Google Scholar]
- Screening and validation of housekeeping genes of the root and cotyledon of Cunninghamia lanceolata under abiotic stresses by using quantitative real-time PCR. Int. J. Mol. Sci.. 2016;17:1998.
- [Google Scholar]
- Genomes (Second ed.). Oxford: Wiley-Liss; 2002. p. :600.
- Quantification of the detrimental effect of a single primer-template mismatch by real-time PCR using the 16S rRNA as an example. Appl. Environ. Microbiol.. 2008;74:1660-1663.
- [Google Scholar]
- Similar sequence-free amplification of human glyceraldehyde-3-phosphate dehydrogenase for real time RT-PCR applications. Mol. Cell. Probes. 2005;19:181-186.
- [Google Scholar]
- Reference genes for RT-qPCR analysis of environmentally and developmentally regulated gene expression in alfalfa. Am. J. Plant Sci.. 2015;6:132-143.
- [Google Scholar]
- Subunit structure of higher plant glyceraldehyde-3-phosphate dehydrogenases (EC 1.2.1.12 and EC 1.2.1.13) J. Biol. Chem.. 1979;254:6094-6098.
- [Google Scholar]
- The effects of internal primer template mismatches on RT-PCR: HIV-1 model studies. Nucleic Acids Res.. 1997;25:654-658.
- [Google Scholar]
- Base Mispair Extension Kinetics: Binding of avian myeloblastosis reverse transcriptase to matched and mismatched base pair termini. J. Biol. Chem.. 1992;4:2633-2639.
- [Google Scholar]
- Nucleotide sequence of the glyceraldehyde-3-phosphate dehydrogenase gene from the mesophilic methanogenic archaebacteria Methanobacterium bryantii and Methanobacterium formicicum. Comparison with the respective gene structure of the closely related extreme thermophile Methanothermus fervidus. Eur. J. Biochem.. 1989;179:405-413.
- [Google Scholar]
- PriFi: using a multiple alignment of related sequences to find primers for amplification of homologs. Nucleic Acids Res.. 2005;33 Web Server issue, W516-W520
- [Google Scholar]
- Processed pseudogenes interfere with reverse transcriptase polymerase chain reaction controls. Anal. Biochem.. 1996;237:157-159.
- [Google Scholar]
- RT-PCR for the pseudogene-free amplification of the gluceraldehyde-3-phosphate dehydrogenase gene (gapd) Mol. Cell. Probes. 2003;17:261-265.
- [Google Scholar]
- Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. U.S.A.. 1992;89:10915-10919.
- [Google Scholar]
- Exploring valid reference genes for gene expression studies in Brachypodium distachyon by real-time PCR. BMC Plant Biol.. 2008;8:112.
- [Google Scholar]
- Hemi-nested touchdown PCR combined with primer-template mismatch PCR for rapid isolation and sequencing of low molecular weight glutenin subunit gene family from a hexaploid wheat BAC library. BMC Genet.. 2007;8:18.
- [Google Scholar]
- mRNA-specific reverse transcription-polymerase chain reaction from human tissue extracts. Anal. Biochem.. 2002;307:304-315.
- [Google Scholar]
- Comparison of reference genes for quantitative real-time polymerase chain reaction analysis of gene expression. Plant Mol. Biol. Rep.. 2004;22:325-337.
- [Google Scholar]
- The effect of multiple primer-template mismatches on quantitative PCR accuracy and development of multi-primer set assays for accurate quantification of pcrA gene sequence variants. J. Microbiol. Methods. 2013;94:224-231.
- [Google Scholar]
- Debate round table on RT-PCR controls: concluding remarks and mini-review-current recommendations for positive controls in RT-PCR assays. Leukemia. 2001;15:1033-1037.
- [Google Scholar]
- Comprehensive analysis of the pseudogenes of glycolytic enzymes in vertebrates: the anomalously high number of GAPDH pseudogenes highlights a recent burst of retrotrans-positional activity. BMC Genomics. 2009;10:480.
- [Google Scholar]
- CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res.. 2007;35:D237-D240.
- [Google Scholar]
- Prokaryotic features of a nucleus-encoded enzyme. Eur. J. Biochem.. 1986;159:323-331.
- [Google Scholar]
- The housekeeping gene glyceraldehyde-3-phosphate dehydrogenase is inappropriate as internal control in comparative studies between skin tissue and cultured skin fibroblasts using Northern blot analysis. Arch. Dermatol. Res.. 1999;291:659-661.
- [Google Scholar]
- Comparison of methods for searching protein sequence databases. Protein Sci.. 1995;4:1145-1160.
- [Google Scholar]
- Searching databases of conserved sequence regions by aligning protein multiple-alignments. Nucleic Acids Res.. 1996;24:3836-3845.
- [Google Scholar]
- MFEprimer-2.0: a fast thermodynamics-based program for checking PCR primer specificity. Nucleic Acids Res.. 2012;40:W205-W208.
- [Google Scholar]
- Amplification of an orthology of a vacuolar anion transporter gene (AtCLC-c) from Lepidium sativum L. J. Phytol. Res.. 2013;26:59-66.
- [Google Scholar]
- Consensus-degenerate hybrid oligonucleotide primers for amplification of distantly related sequences. Nucleic Acids Res.. 1998;26:1628-1635.
- [Google Scholar]
- Effect of experimental treatment on housekeeping gene expression: validation by real-time, quantitative RT-PCR. J. Biochem. Biophys. Methods. 2000;46:69-81.
- [Google Scholar]
- GAPDH: Biological Properties and Diversity. Advances in Experimental Medicine and Biology 985. Dordrecht: Springer Science+Business Media; 2003. pp. 295
- Finding sequence motifs in groups of functionally related proteins. Proc. Natl. Acad. Sci. U.S.A.. 1990;87:826-830.
- [Google Scholar]
- The effect of primer-template mismatches on the detection and quantification of nucleic acids using 5’ nuclease assay. J. Mol. Diagn.. 2010;12:109-117.
- [Google Scholar]
- Pseudogenes as weaknesses of ACTB (Actb) and GAPDH (Gapdh) used as reference genes in reverse transcription and polymerase chain reaction. PLoS ONE. 2012;7:e41659.
- [Google Scholar]
- Housekeeping genes as internal standards: use and limits. J. Biotech.. 1999;75:291-295.
- [Google Scholar]
- CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weigh matrix choice. Nucleic Acids Res.. 1994;22:4673-4680.
- [Google Scholar]
- Sequence variation in primer targets affects the accuracy of viral quantitative PCR. J. Clin. Virol.. 2005;34:104-107.
- [Google Scholar]
- Quantitative effects of position and type of single mismatch on single base primer extension. J. Microbiol. Methods. 2009;77:267-275.
- [Google Scholar]
- Selection of suitable reference genes for quantitative real-time PCR gene expression analysis in salix matsudana under different abiotic stresses. Sci. Rep.. 2017;7:40290.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.jksus.2017.07.004.
Appendix A
Supplementary data
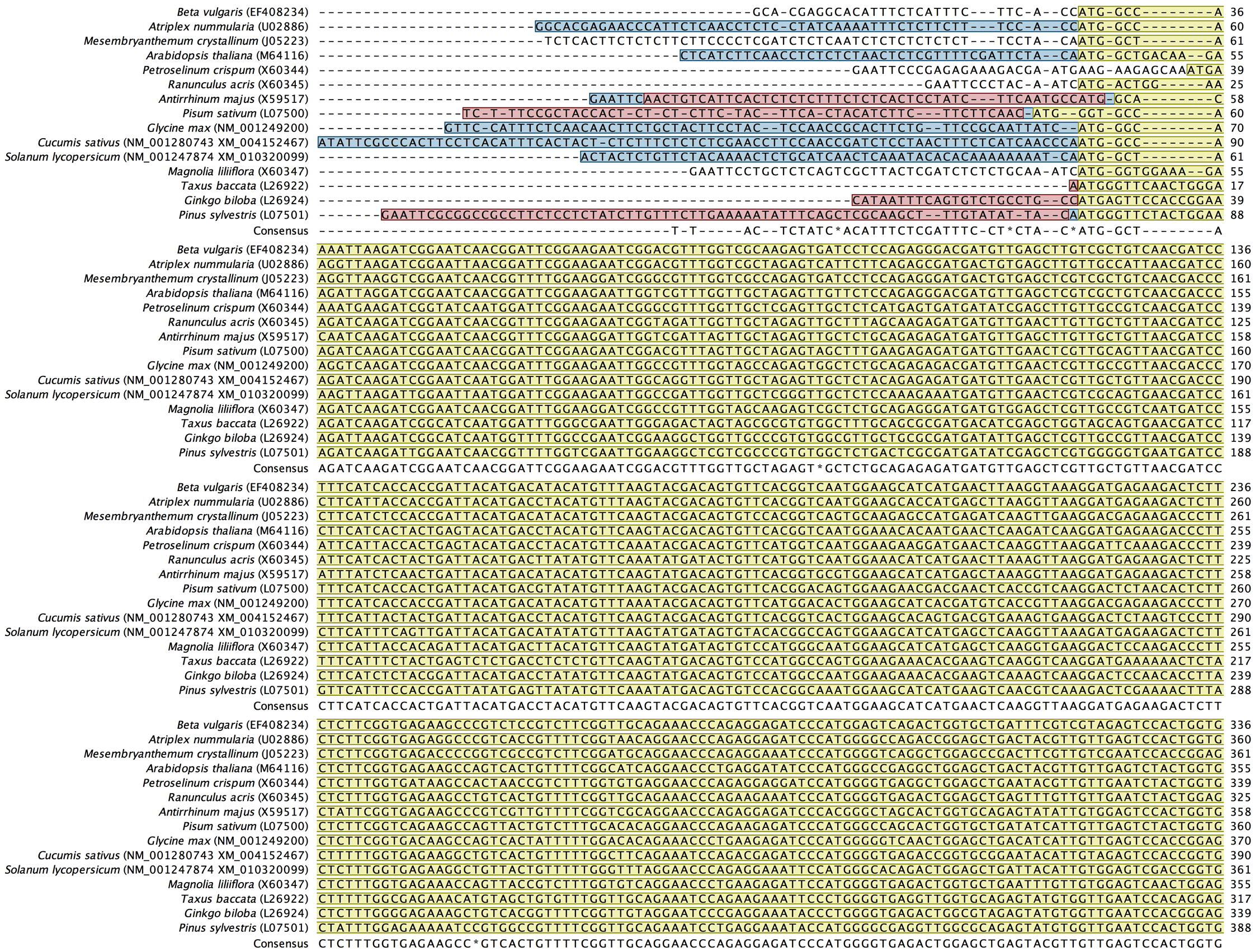
Alignment of back-translated DNA sequences of homologous orthologs GAPDH protein coding regions. Alignments were generated using ClustalW. Gaps have been introduced to permit alignment. Coding sequences are highlighted with light brown color. Blue highlighted sequences indicate the remaining part of each gene, and purple highlighted sequences indicate the 5′ and 3′ end sequences. Species included in the alignment and their accession numbers (between brackets) are shown beside each corresponding sequence.
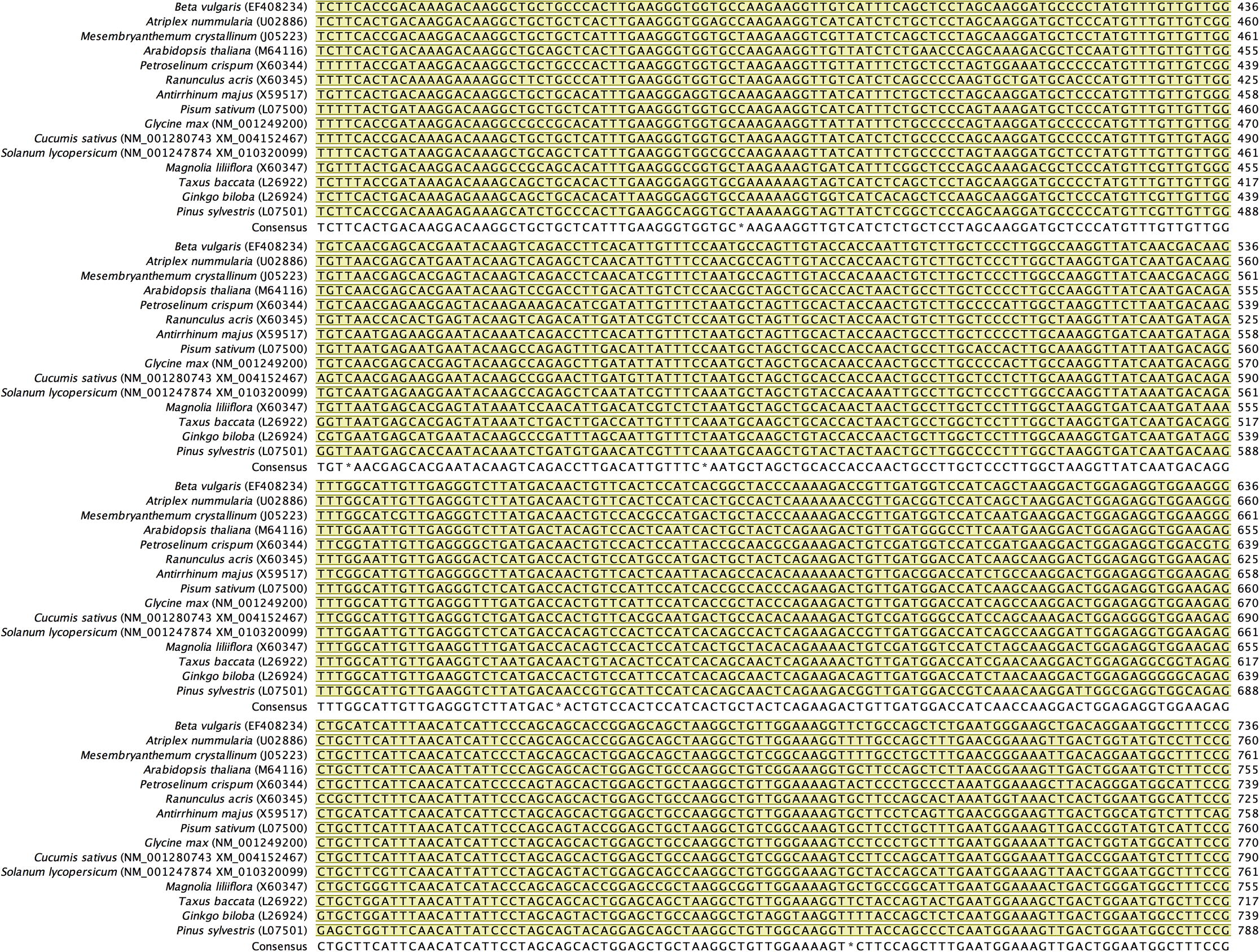
Alignment of back-translated DNA sequences of homologous orthologs GAPDH protein coding regions. Alignments were generated using ClustalW. Gaps have been introduced to permit alignment. Coding sequences are highlighted with light brown color. Blue highlighted sequences indicate the remaining part of each gene, and purple highlighted sequences indicate the 5′ and 3′ end sequences. Species included in the alignment and their accession numbers (between brackets) are shown beside each corresponding sequence.
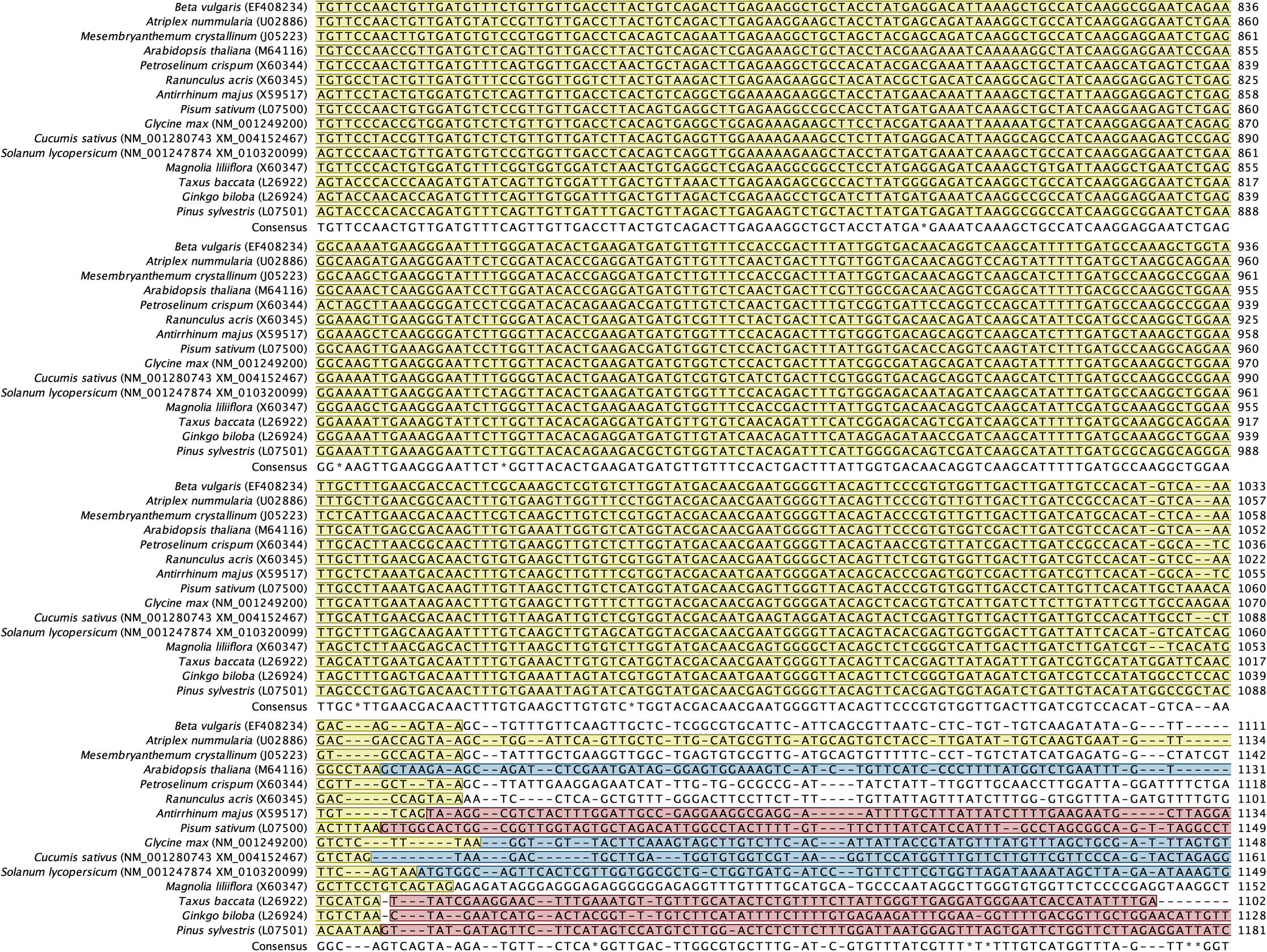
Alignment of back-translated DNA sequences of homologous orthologs GAPDH protein coding regions. Alignments were generated using ClustalW. Gaps have been introduced to permit alignment. Coding sequences are highlighted with light brown color. Blue highlighted sequences indicate the remaining part of each gene, and purple highlighted sequences indicate the 5′ and 3′ end sequences. Species included in the alignment and their accession numbers (between brackets) are shown beside each corresponding sequence.
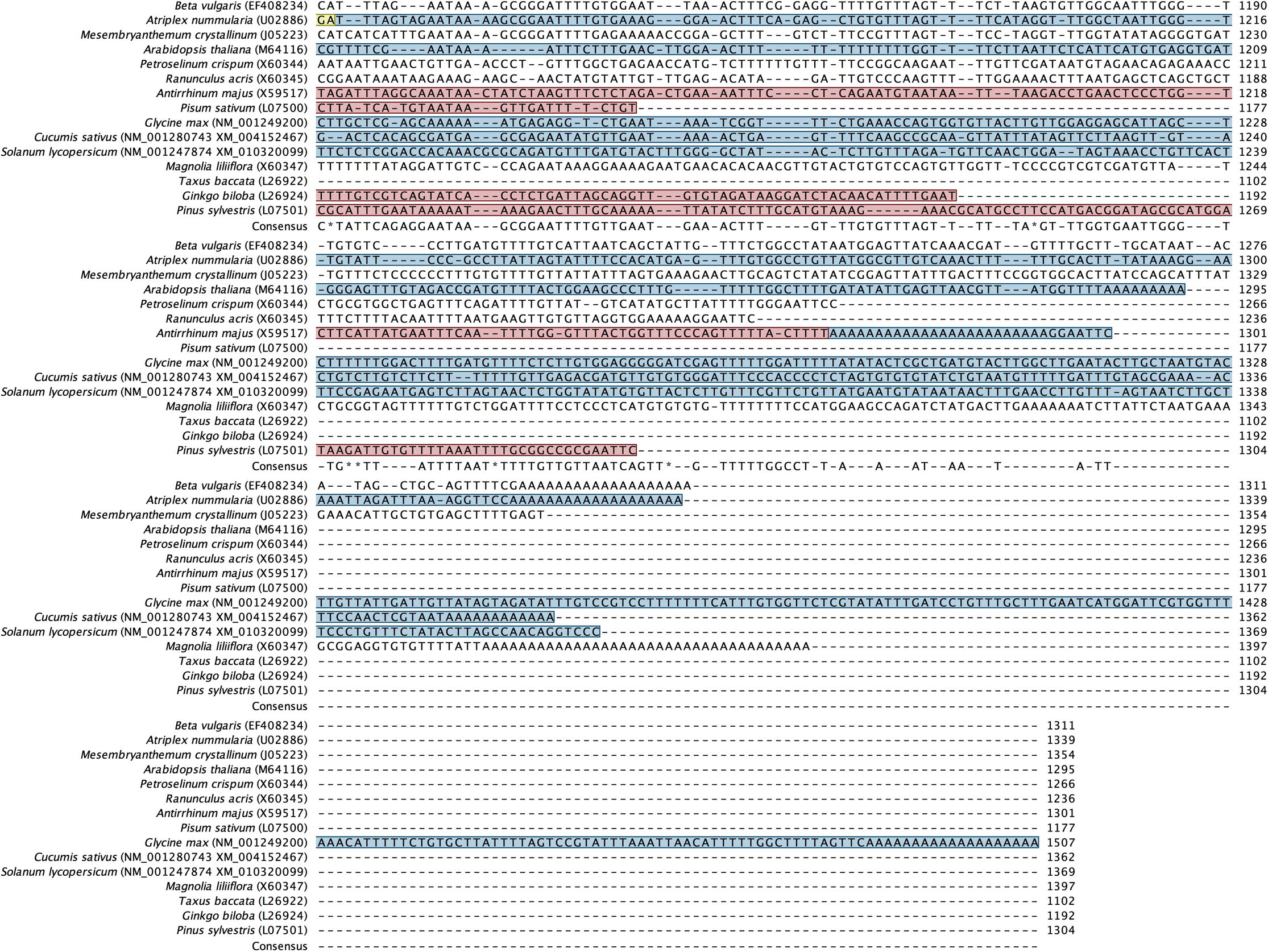
Alignment of back-translated DNA sequences of homologous orthologs GAPDH protein coding regions. Alignments were generated using ClustalW. Gaps have been introduced to permit alignment. Coding sequences are highlighted with light brown color. Blue highlighted sequences indicate the remaining part of each gene, and purple highlighted sequences indicate the 5′ and 3′ end sequences. Species included in the alignment and their accession numbers (between brackets) are shown beside each corresponding sequence.




