

Translate this page into:
Integrating RNA-seq and machine learning to identify novel biotargets and high-affinity ligands for cardiovascular disease management
* Corresponding author E-mail addresses: sabiha-khatoon@ouhsc.edu (S. Khatoon), shhaque@jazanu.edu.sa (S. Haque)
-
Received: ,
Accepted: ,
Abstract
Cardiovascular diseases (CVDs) are the leading cause of mortality globally and, due to their heterogeneous nature, present significant clinical challenges. This study aims to identify novel biotargets for CVDs and propose potential inhibitors against them. The study leverages RNA-sequencing data in conjunction with machine learning (ML) techniques to uncover differentially expressed genes (DEGs) as potential biotargets for CVDs. Transcriptomic data was obtained from the Gene Expression Omnibus (GEO) database, and DESeq2 was used to identify DEGs. Machine learning (ML) models, random forest (RF), and support vector machines (SVM) were used to characterize DEGs and to rank top genes as biomarkers. Functional annotation of top hub genes was performed using clusterProfiler and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology (GO) analyses. Protein-protein interaction (PPI) networks were constructed using STRING. Molecular docking analyses were conducted using Biovia Discovery Studio and AutoDock Vina, targeting top genes with ligands sourced from the Drug Gene Interaction Database as repurposable targets. Comprehensive analysis of DEGs led to the identification of multiple hub genes and predictive biomarkers for CVD treatment. Using ML algorithms for biomarker feature prediction, we identified the top DEGs, which included interleukin-6 (IL6), tumor necrosis factor (TNF), myosin heavy chain-6 (MYH6), apolipoprotein E (APOE), low-density lipoprotein receptor (LDLR), proprotein convertase subtilisin/kexin type-9 (PCSK9), angiotensin-converting enzyme (ACE), actin alpha-2 (ACTA2), activated protein kinase (AMP)-activated non-catalytic subunit γ-2 (PRKAG2), and cardiac type troponin T2 (TNNT2). Network and PPI analyses further highlighted the significance of the identified DEGs, which were then targeted for discernment of high-affinity binding ligands from clinically approved and relevant drugs using docking studies. Biomarker-guided approaches for the prediction, evaluation, diagnosis, and treatment of CVDs hold substantial promise for clinical application. The identification of clinically approved ligands targeting the top genes from DEGs in CVD patients might facilitate more effective personalized treatment regimens, improving patient outcomes and ultimately transforming CVD management.
Keywords
Biomarkers
Cardiovascular diseases
Hub genes
Machine learning
RNA-sequencing

1. Introduction
Cardiovascular diseases (CVDs) represent a wide spectrum of conditions associated with morpho-functional deficits of heart tissues and vessels (Olvera Lopez et al., 2023). CVDs include rheumatic heart disease (RHD), coronary heart disease (CAD), stroke, and heart failure, and pose significant global public health concerns. Progressive increases in the aged populations, coupled with lifestyle changes linked with urbanization and industrialization, have resulted in dramatic rises in incidences of diabetes, hypertension, and obesity, which are significant CVD risk factors (Schnall et al.,2016). According to the World Health Organization (WHO), CVDs are the main cause of mortality worldwide, claiming over 17 million lives annually, with a projected increase to over 23.6 million by 2030 (World Health Organization, 2023), necessitating urgent needs for the discovery of novel ways of early identification and intervention. While there are various drugs recommended for hypertension, hyperlipidemia, and other CVD risk factors, their efficacy varies greatly across subjects. This is both due to etiological heterogeneity and significant gaps in our understanding of the underlying biological mechanisms (American Diabetes Association Professional Practice Committee, 2022). Identification of genes and proteins that are differently regulated in the various classes of CVDs may give deeper mechanistic insights into the malfunctioning pathways (Wang et al., 2017). Further, clarifying therapeutic vulnerabilities via biomarker-guided patient selection regimens will aid in the creation and repurpose of targeted medicines (Dara et al., 2022). Current risk assessment is mostly based on broad, nonspecific markers (age, family history, blood pressure, blood lipids, etc.), which have poor predictive value, particularly in asymptomatic phases (Upadhyay et al., 2015). For efficient diagnoses and management, new biomarkers with high sensitivity and specificity and tailored therapeutic approaches are essential (Vadapalli et al., 2022).
Recent transcriptomic advancements have allowed a more comprehensive characterization of disease-associated gene alterations. One of these technologies, RNA-seq, is a reliable, high-throughput method for detecting differentially expressed genes (DEGs) (Di Salvatore et al., 2023). The functional analysis of disease-specific RNA signatures has the potential to identify the underlying biological pathways and mechanisms of disease pathology, including CVDs (Seo et al., 2006; Ahmed et al., 2021). DEG indicators with clinical significance may aid in improving risk prediction, diagnosis, and individualized treatment for patients (Byron et al., 2016). However, traditional screening of DEGs is restricted in its capacity to identify clinically meaningful signals. Machine learning (ML) techniques that use large-scale multi-omics data can overcome this issue by identifying reliable predictive biomarkers through automated feature selection and categorization (Ahmed et al., 2020). ML algorithms can detect subtle patterns and relationships in data that are often overlooked by traditional statistical approaches (Bostanci et al., 2023). The combination of RNA-seq-based transcriptomics with these techniques allows for the analysis of disease regulation at various biological levels to identify potential therapeutic targets (Casamassimi et al., 2017). Subsequently, molecular docking and simulation may be utilized for structure-based drug design to screen binding candidates against the target proteins involved in disease pathogeneses (Torres et al., 2019, Agu et al., 2023). Further, molecular docking was used to scan FDA-approved drug libraries for molecules with strong binding affinity against protein products of top DEG targets, which might serve as prospective therapeutic lead compounds. In this study, we followed the above outlined integrated multi-omics approach in order to identify pertinent biotargets associated with CVD pathology, and then obtain high-affinity ligands from the clinically approved drugs.
2. Experimental procedures
2.1 Transcriptomic data acquisition
Public transcriptomic database, Gene Expression Omnibus (GEO) (Clough and Barrett 2016) was employed to obtain the list of DEGs. Five GEO datasets (GSE262161, GSE222118, GSE255895, GSE263644, and GSE242046) were used, amounting to 140 samples altogether, including controls (51) and CVD cases (89).
2.2 Data preprocessing and screening for differentially expressed genes/transcripts
RNA-seq data from whole blood samples of CVD patients and healthy controls were explored. ComBat (R package) was utilized to remove batch effects, creating homogeneity in a common gene-based unified dataset (Ritchie et al., 2015). Low-expression genes with less than 10 counts for less than two samples were eliminated before differential expression analysis between subjects and controls. The criterion of selection of upregulated and downregulated genes was log2 fold change > 1 and < -1, respectively. Such DEGs, which were either up-regulated or down-regulated in patient samples compared to controls, were analyzed in order to find potential biomarkers and target genes.
2.3 Identification of top transcriptional signatures using ML feature Selection
To identify the most relevant features (genes) that differentiate CVD patients from controls, both approaches, linear and non-linear relationships were used. Linear and non-linear relationships were assessed using support vector machine (SVM) and random forest (RF), respectively, which were fine-tuned by stratified k-folds cross-validation to avoid overfitting of the model. Coefficients derived from the SVM were employed to rank the importance of individual features. The model identified 100 features based on their relevance. Cross-validation was fundamental in estimating the performance of models to classify unseen data, thus reducing overfitting. Further, the methodology evaluated the performance of the classifier against benchmark classification metrics such as accuracy, precision, recall, F1, area under the curve (AUC), and receiver operating characteristic (ROC). For non-linear relationships between the features, tree-based algorithm, RF was utilized to account for more complex interactions between features. The models were tested against the same metrics so that both linear and non-linear relationships were pursued in order to achieve best performance (Akinnuwesi et al., 2023).
2.4 Protein-protein interaction network construction and identification of hub genes
To get mechanistic insights of CVD-related DEGs, a protein-protein interaction (PPI) network was constructed using the STRING platform. The PPI network was further evaluated using Cytoscape software (Majeed and Mukhtar, 2023), with the CytoHubba plugin used for identification of strongly linked hub genes in the network. The major hub genes were identified using the maximal clique centrality (MCC) method (Li and Xu, 2019), which considers both direct and indirect relationships.
2.5 Identification of common transcriptional signatures and pathway and function enrichment analyses for CVDs
Top hub DEGs related to CVD pathology were used to identify shared transcriptional regulators, pathways, and CVD markers. Functional pathway enrichment analysis of the shared genes was carried out using computational methods like clusterProfiler (R package) (Wu et al., 2021), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Gene Ontology (GO) to acquire biological understanding of the pathophysiological processes underlying the disease. Biological processes, molecular functions, and cellular components were used to categorize GO keywords. Signaling pathways and activities that are affected by cardiovascular pathology were discovered using KEGG pathway analysis. To validate enriched keywords, Gene Set Enrichment Analysis (GSEA) was carried out on whole transcriptome profiles, with an adjusted p-value of < 0.05 deemed as substantially enriched.
2.6 Virtual screening and molecular docking
Potential pharmacological inhibitors targeting DEGs implicated in CVDs were identified by molecular docking. Protein Data Bank (PDB) was employed for the retrieval of 3D structures of the identified hub proteins. Given their established modes of action against inflammatory and other disorders, only clinical approved medicinal compounds from the Drug Gene Interaction Database (DGIdb) were screened for ligand-binding activities based on the drug gene interaction score. Biovidia Computational Biology tool (Discovery Studio) was used to complete the docking steps. Grid spacing of 1 Å was used for the identified hub proteins, IL6 and TNF because of their smaller binding pockets. Because the binding cleft is relatively larger in size for MYH6, a wider grid box and more space between cells was specified. Flexible docking, which allows for the movement of the rotatable bonds in ligands, was followed to get the best binding conformations, and the best positions were determined according to Vina’s binding affinity ratings.
3. Results
3.1 Differential expression profiles in CVDs
Unified RNA-seq GEO datasets based on 23,454 common genes were used to profile the gene expression of samples from CVD clinical cases, in comparison to controls. DESeq2 analysis identified 1205 genes that were differently expressed between cases versus controls, with 678 up-regulated and 527 down-regulated genes (Fig. 1).
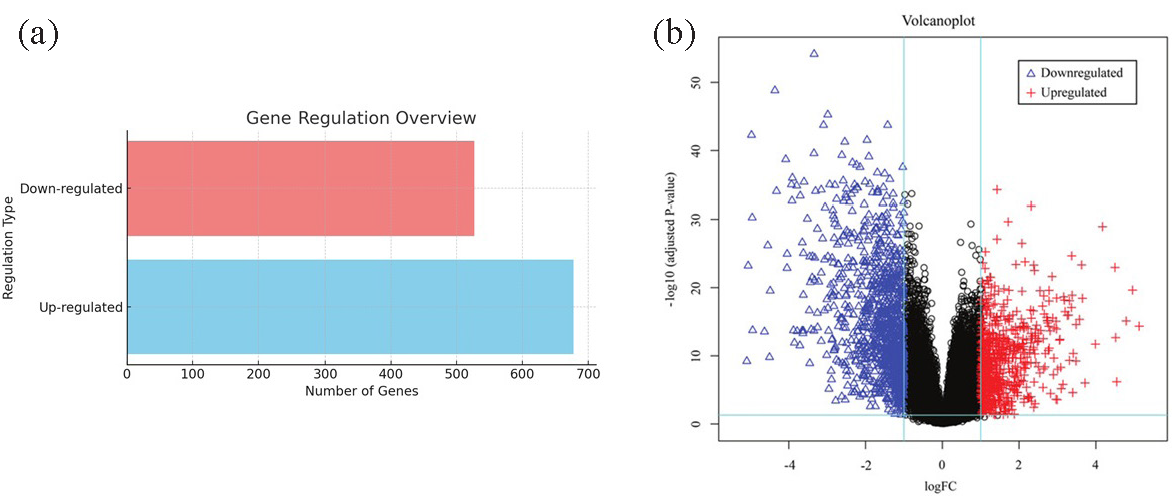
- Identification of DEGs in CVD subjects, compared to controls. (a) Number of up-regulated DEGs was 678, while there were 527 DEGs which were down-regulated. (b) Differential gene expression between the two groups is depicted as a volcano plot, depicted as blue triangles (downregulated) and red crosses (upregulated). LogFC: Log fold change, DEG: Differentially expressed genes, CVD: Cardiovascular disease.
3.2 Cluster analyses of DEGs
Hierarchical clustering was used to categorize DEGs based on expression patterns across all data. Genes were categorized using hierarchical clustering to discover probable disease categories based on gene expression patterns. To identify enriched areas, the genomic distribution of DEGs was examined, and the key driver genes were chosen based on their connectedness in weighted gene co-expression networks. In Fig. 2(a), scale independence is represented as R2 against different levels of power of soft thresholding. Mean connectivity in Fig. 2(b) decreased with increase in power, which is characteristic of scale-free networks. Connectivity histogram of k as depicted in Fig. 2(c) indicated that most genes have low connectivity, while very few genes were highly connected. Such right-skewed distribution is generic for biological networks. Log-log plot of connectivity, showing the network has been depicted in Fig. 2(d). Given R2 (0.87) and a slope close to-1.3, the curve shows a fair fit to be scale-free in nature. Distinct clusters of co-expressed genes were discovered through identifying the optimal soft thresholding power for building a weighted gene co-expression network in Weighted Gene Correlation Network Analysis (WGCNA) based on 140 samples.
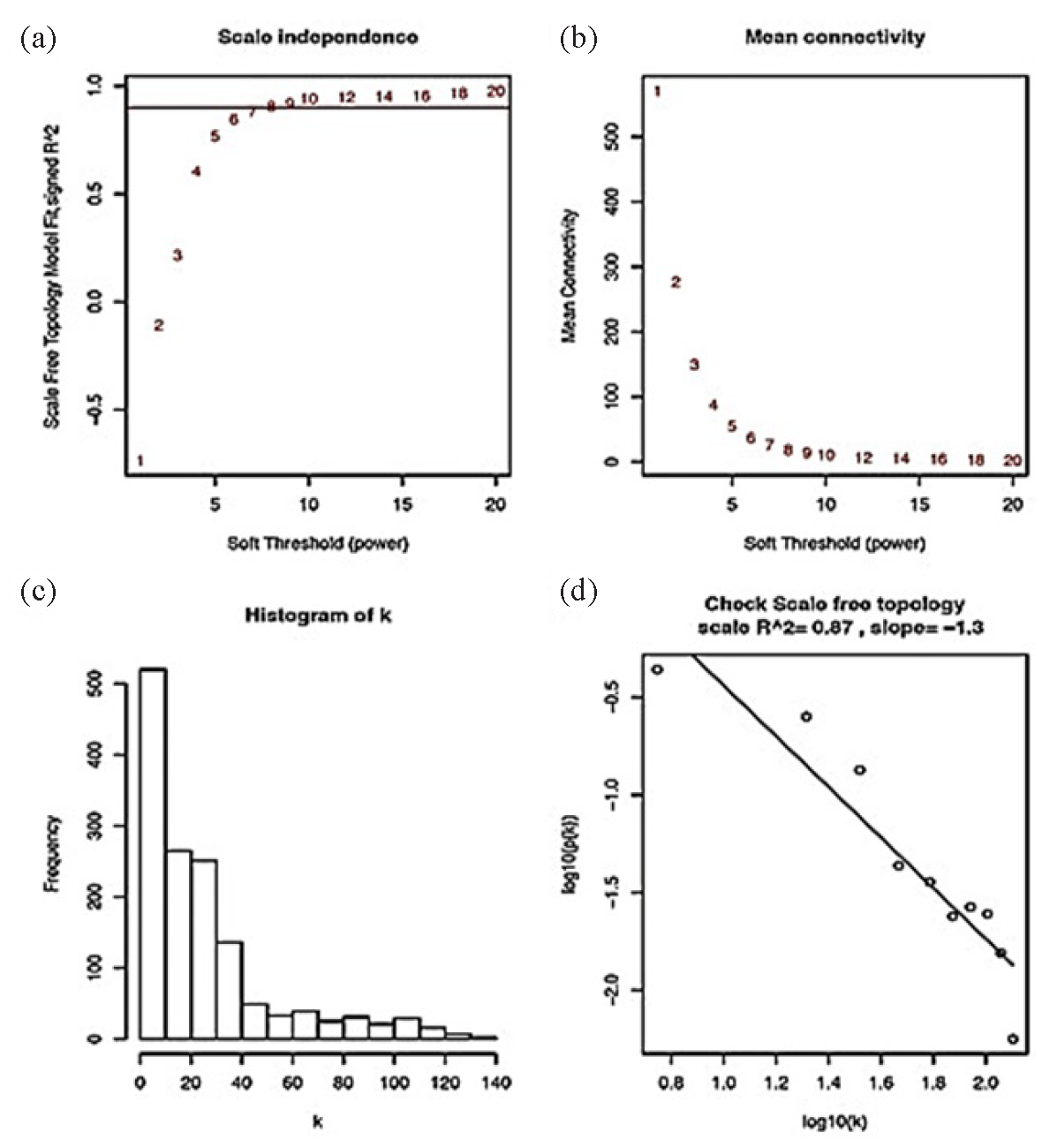
- (a-d) Scale-free fit index for different soft-thresholding powers, displaying the correlation coefficients of log(k) and log(p(k)) corresponding to various soft thresholds, and represents the mean values of the gene adjacency coefficients according to various soft thresholds, which represent the network’s average degree of connectedness.
3.3 Feature selection using ML
SVM and RF classifiers identified the significant biomarkers to differentiate CVD patients from controls, and classified performance, feature importance, and overlap in selected features. Fig. 3(a) presents the ROC curves of the two classifiers, showing that SVM attains a higher AUC value at 0.88, compared to RF at 0.83, indicating that SVM is better in distinguishing CVD patients from healthy controls. As shown in the precision-recall comparison (Fig. 3b), SVM constantly achieved a higher precision at almost all recall levels, making it more efficient for identifying true positives in imbalanced datasets. A cross-validation metrics heatmap further confirmed the dominance of SVM’s stronger classification ability over RF through all metrics, particularly for the ROC AUC values (Fig. 3c).
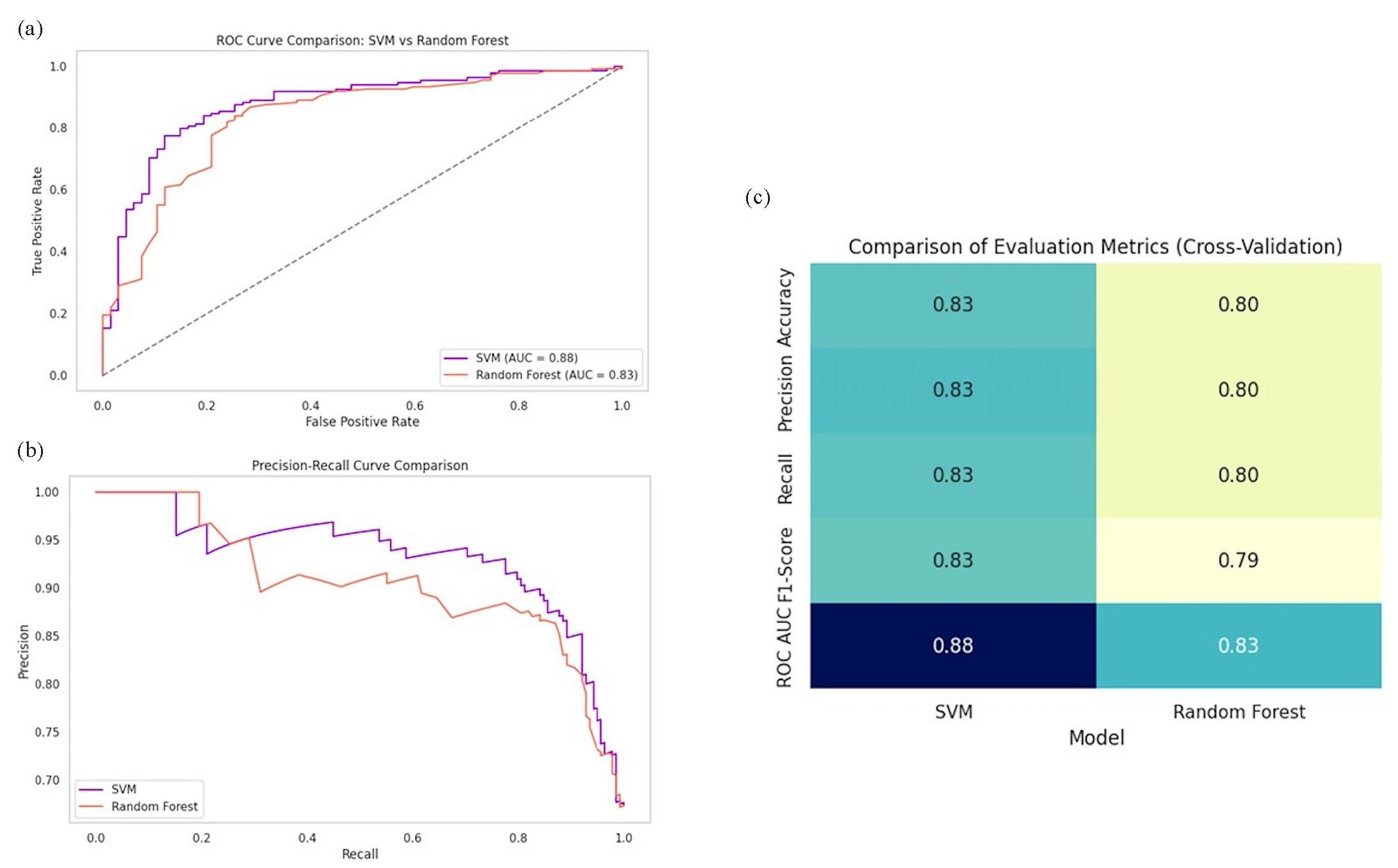
- Comparison of support vector machine (SVM) and random forest performance. (a) ROC curve with SVM (AUC=0.88) outperforming random forest (AUC=0.83). (b) Precision-recall curve highlights SVM’s superior precision. (c) Heatmap compares cross-validation metrics, confirming SVM’s superior performance.
3.4 Pathway enrichment analyses
GO pathway analyses were utilized to characterize the pathophysiological relevance of the identified CVD-associated DEGs (Fig. 4), highlighting important molecular and biological processes of CVDs and emphasizing roles for lipid metabolism, inflammation, and vascular remodeling. Molecular functions that were enriched with high statistical confidence, such as binding to the LDL particle, sterol transfer activity, and binding to the TNF receptor, underline the role of dyslipidemia and inflammatory signaling in CVDs. Biological processes, such as regulation of lipid storage, inflammatory responses, and cell motility, further connect metabolic and vascular dysfunctions to CVD progression. Pathway enrichment, including advanced glycation end products- receptor for advanced glycation end products (AGE-RAGE) signaling, cholesterol metabolism, and lipid and atherosclerosis pathways, connect systemic inflammation, diabetes-related complications, and metabolic syndromes to CVDs.
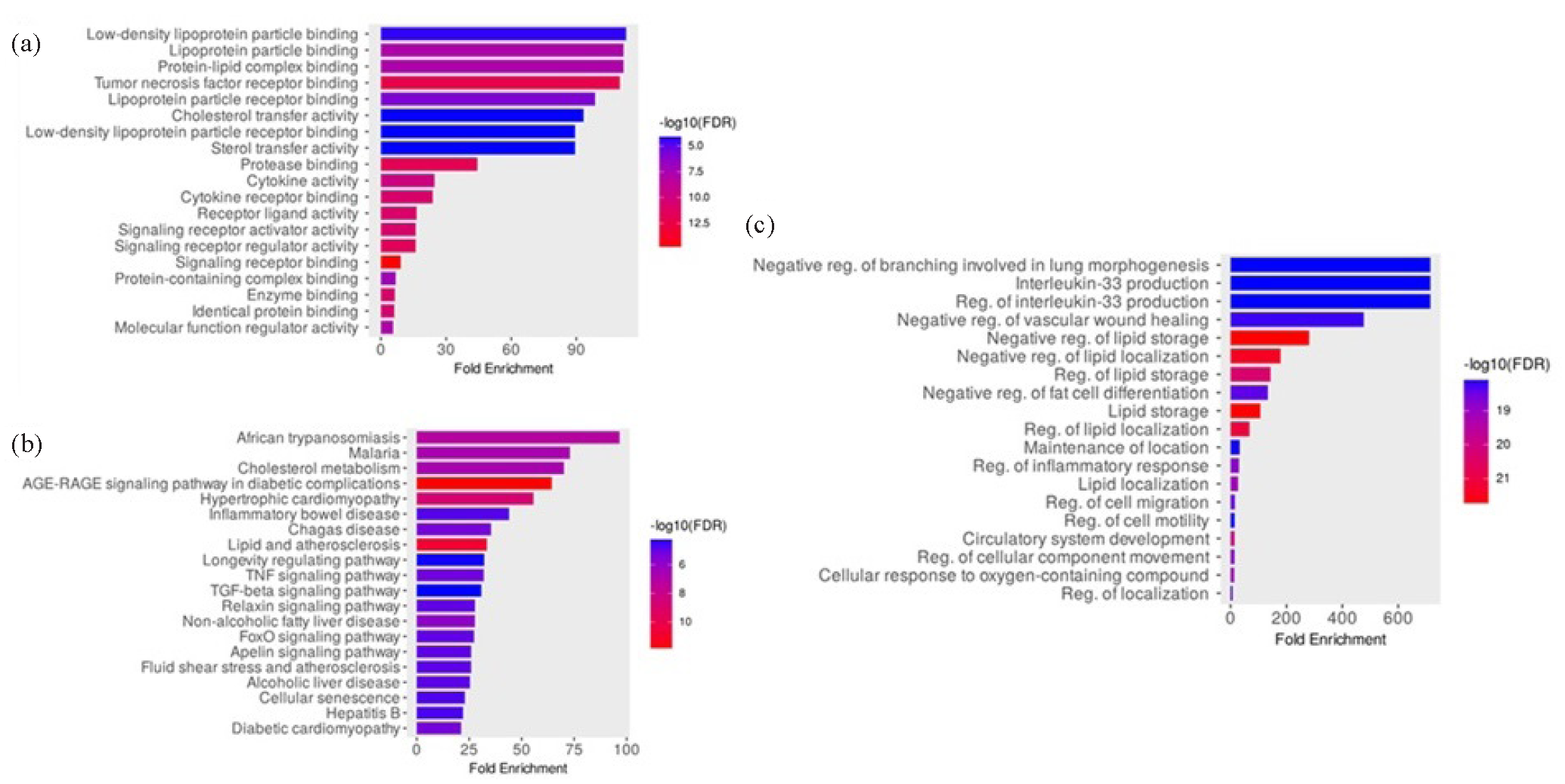
- DEGs’ GO enrichment analysis and module identification. The most significant GO molecular function enrichment terms for the DEGs associated with CVD pathology are displayed in (a-c) display the top KEGG pathway modules and GO biological process terms, respectively, associated with the clinical status of CVDs. DEGs: Differentially expressed genes, GO: Gene ontology, CVD: Cardiovascular disease.
3.5 PPI network and hub genes
Gene protein interactomes were found and PPI network was constructed on the STRING platform, which confirmed coordinated dysregulation of biological processes in CVDs (Supplementary File S1). Functional relationships between co-expressed genes were examined, and interleukin-6 (IL6), tumor necrosis factor (TNF), myosin heavy chain-6 (MYH6), apolipoprotein E (APOE), low-density lipoprotein receptor (LDLR), proprotein convertase subtilisin/kexin type-9 (PCSK9), angiotensin-converting enzyme (ACE), actin α-2 (ACTA2), activated protein kinase (APM)-activated non-catalytic subunit gamma-2 (PRKAG2), and cardiac type troponin T2 (TNNT2) were identified as significant hub genes using CytoHubba plugin (Table 1; Fig. 5). Integrated enrichment analysis of GO terms related to CVDs focusing on pathways, molecular functions, and biological processes showed the enriched pathways such as apelin signaling pathways involved in cardiovascular function and angiogenesis and AGE-RAGE signaling pathways associated with diabetic complications and vascular inflammation (Fig. 6a). Other important pathways found related to TNF signaling, associated with inflammation and atherosclerosis, cholesterol metabolism, and longevity-regulating pathway, implying that lipid dynamics, in conjunction with aging, may play significant roles in cardiovascular health. Fig. 6(b) represents the significantly enriched molecular functions of hub genes, which included functions such as binding to LDL particles, important in lipid metabolism, and the central activity linked to atherosclerosis and cytokine activity to regulate inflammatory responses implicated in CVD progression. Additionally, cholesterol transfer activity is found important for the lipid transport processes and formation of plaques in atherosclerosis. Biological processes identified (Fig. 6c) included such as the regulation of lipid localization, which is important for lipid transport and storage, as well as the production of interleukin-33 (IL-33) which is the major mediator in vascular repair and inflammation. Involvement of processes such as regulation of differentiation in fat cells as well as modulation of inflammatory response highlighted that there are other interactions of metabolic and inflammatory mechanisms, which further drive CVDs. Overall, all the interacting networks represented in Fig. 6 confirm the intricately complex and multifactorial etiology of CVDs involving a plethora of pathways, molecular functions, and biological processes.
| Gene name | CVDs dataset | |
|---|---|---|
| (log2FC) | P-value | |
| IL6 | 1.2 | 0.07 |
| TNF | 1.3 | 0.08 |
| MYH6 | 1.4 | 0.06 |
| APOE | 1.1 | 0.09 |
| LDLR | 1.5 | 0.07 |
| PCSK9 | 1.2 | 0.1 |
| ACE | 1.3 | 0.09 |
| ACTA2 | 1.4 | 0.08 |
| PRKAG2 | 1.2 | 0.07 |
| TNNT2 | 1.3 | 0.08 |
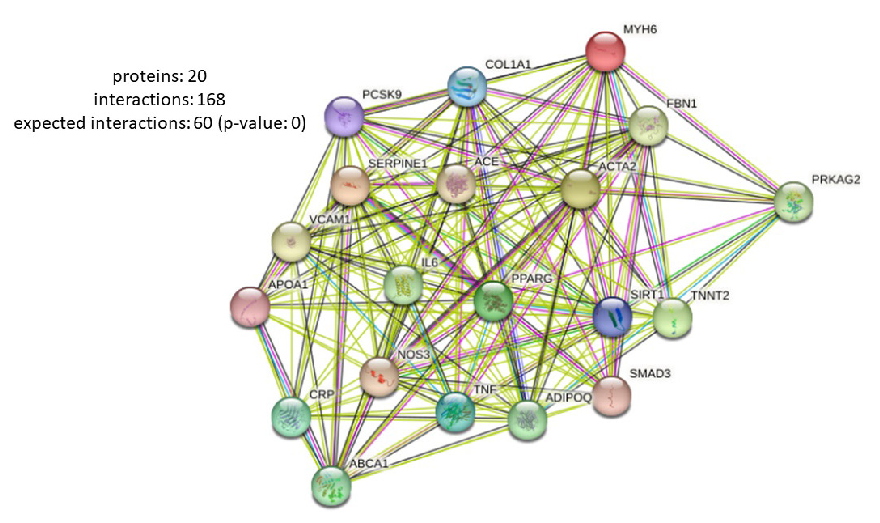
- PPI analysis of the hub DEGs associated with CVD pathogenesis obtained using STRING database. PPI: Protein-Protein interaction, DEG: Differentially expressed genes, CVD: Cardiovascular disease.
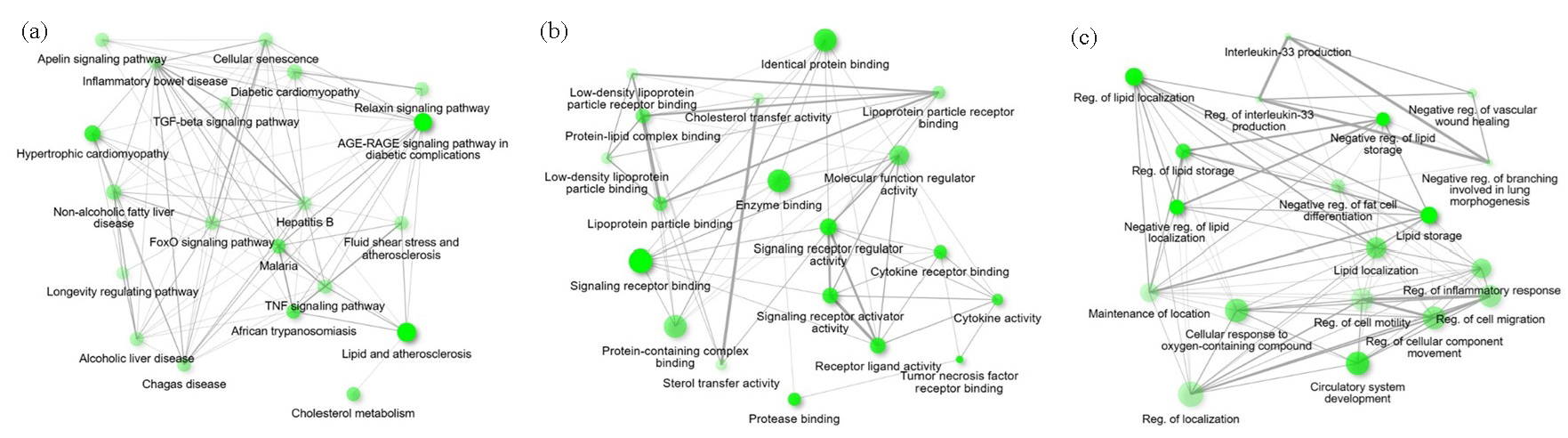
- Functional analysis of the hug DEGs using ClueGO. (a) GO biological process (BP) analysis; (b) GO cellular component (CC) analysis and (c) GO molecular function (MF) analysis. DEG: Differentially expressed genes, GO: Gene ontology.
3.6 Molecular docking
Molecular docking using Biovia Discovery Studio was employed for the identification of high-affinity ligands against the hub genes associated with DEGs in CVD cases (Figs. 7 and 8). The 3D structures of IL6, TNF, MYH6, APOE, LDLR, PCSK9, ACE, ACTA2, PRKAG2, and TNNT2 were obtained from PDB. To find possible ligands with known mechanisms of action against inflammatory, metabolic and cardiovascular ailments, DGIdb database was searched for authorized medication compounds. For docking, grid box size and spacing were determined by active site features of the targets. Flexible docking allowed ligand rotations for the identification of optimal binding conformations. The best target-ligand combinations were highlighted based on their binding affinities scored in kcal/mol, as assessed by AutoDock Vina (Table 2). Thus, IL6 presented a docking score of -8.5 kcal/mol (prediction score of 0.81) with evofolisat. TNF showed interaction with rosuvastatin (-9.1 kcal/mol; prediction score 0.76), MYH6 with atenolol (-8.2 kcal/mol), and APOE with pitavastatin (-7.9 kcal/mol; prediction score 0.68). Similarly, LDLR was found to interact with ezetimibe (-8.3 kcal/mol; prediction score 0.71), and PCSK9 with alirocumab (-8.7 kcal/mol; prediction score 0.78). ACE had a binding energy change of -8.9 kcal/mol with lisinopril (prediction score 0.79), while ACTA2-verapamil binding was associated with -7.6 kcal/mol, with a prediction score of 0.65. Binding of metformin with PRKAG2 involved energy change of -7.2 kcal/mol (prediction score 0.62), while binding of omecamtiv mecarbil to TNNT2 had an energy change of -8.1 kcal/mol and a prediction score of 0.69. The results indicate new ligand-protein interactions that may be beneficial for the designing of therapeutic drugs against CVDs.
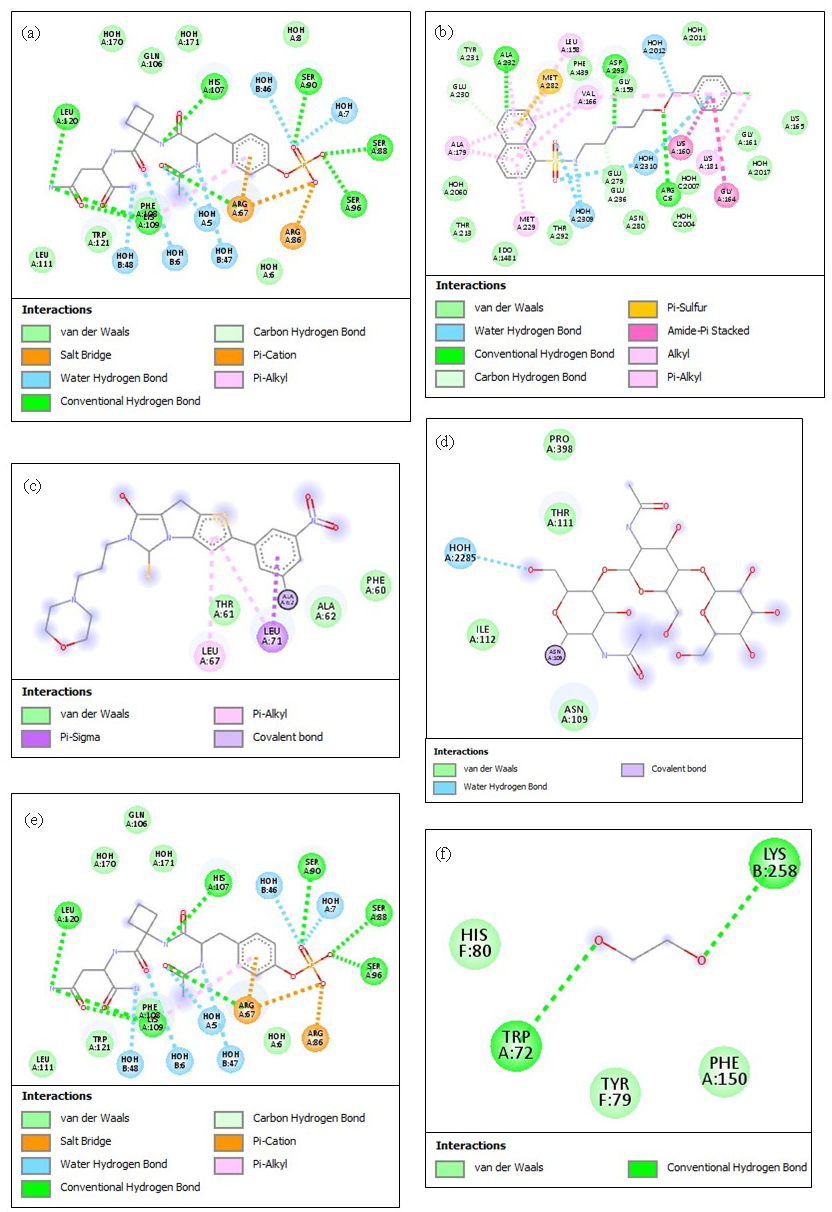
- The 2D illustrations display the best docking positions and the interactions between (a) IL6 and evofolisat, (b) TNF and rosuvastatin, (c) MYH6 and atenolol, (d) APOE and pitavastatin, (e) LDLR and ezetimibe, and (f) PCSK9 and evofolisat. The different types of interactions are depicted in a color-coded manner for the respective panels. TNF: Tumor Necrosis Factor, MYH6: Myosin Heavy Chain 6, APOE: Apolipoprotein E, LDLR: Low-Density Lipoprotein Receptor.
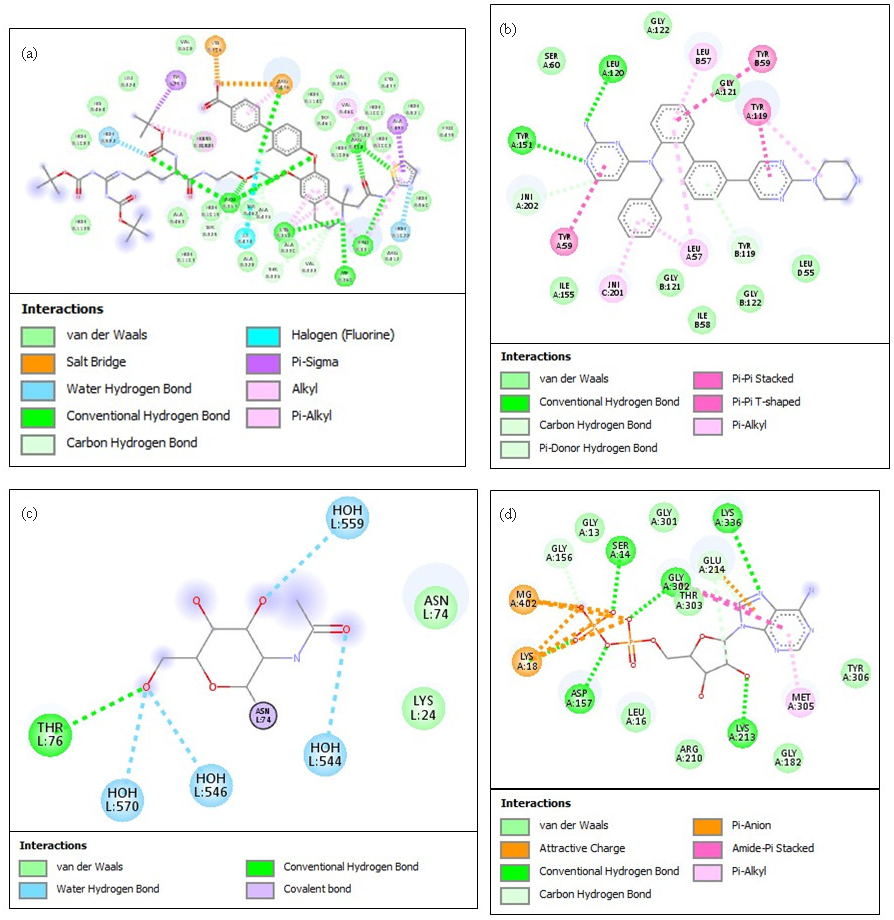
- Best docking positions and the interactions between top hub DEGs and their identified potentially repurposable ligands. (a) The ligand-protein binding is depicted for ACE-lisinopril, (b) ACTA2-verampamil, (c) PRKAG2-metformin, and (d) TNNT2-omecamtiv mecarbil complexes. The various types of interactions are depicted in a color-coded manner for the respective panelsv mecarbil (d) complexes. The various types of interactions are depicted in a color-coded manner for the respective panels. DEG: Differentially Expressed Genes, ACE: Angiotensin-Converting Enzyme, ACTA2: Actin Alpha Cardiac Muscle 2, PRKAG2: Protein Kinase AMP-Activated Non-Catalytic Subunit Gamma 2, TNNT2: Troponin T2, Cardiac Type.
| Target protein | Ligand | Docking score (kcal/mol) | Swiss target prediction score |
|---|---|---|---|
| IL6 | Evofolisat | -8.5 | 0.81 |
| TNF | Rosuvastatin | -9.1 | 0.76 |
| MYH6 | Atenolol | -8.2 | 0.72 |
| APOE | Pitavastatin | -7.9 | 0.68 |
| LDLR | Ezetimibe | -8.3 | 0.71 |
| PCSK9 | Alirocumab | -8.7 | 0.78 |
| ACE | Lisinopril | -8.9 | 0.79 |
| ACTA2 | Verapamil | -7.6 | 0.65 |
| PRKAG2 | Metformin | -7.2 | 0.62 |
| TNNT2 | Omecamtiv mecarbil | -8.1 | 0.69 |
4. Discussion
In order to find biomarkers and therapeutic targets for CVDs which is a heterogeneous group of pathological conditions, RNA-seq gene expression data was combined with ML approaches, which may enhance the possibility for accurate diagnosis, individualized care, and enhanced patient outcomes (Doran et al., 2021). Our analyses revealed pertinent DEGs associated with CVDs, identifying 678 upregulated and 527 downregulated genes. By identifying specific gene clusters with distinctive expression patterns, one could distinguish between tissues that have been impacted by CVD and healthy ones. These discoveries may provide knowledge of the molecular subtypes of CVDs, which is essential for individualized diagnosis and therapy (Leopold et al., 2020). These combined clusters’ gene expressions, which were relevant to CVD, were extracted and transposed, to add labels of 0 for normal and 1 for CVD patients. Further analysis revealed IL6, TNF, MYH6, APOE, LDLR, PCSK9, ACE, ACTA2, PRKAG2, and TNNT2 as hub DEGs and potential biomarkers and therapeutic targets for CVDs. Amongst these, IL6, TNF, MYH6, and APOE showed an up-regulated expression in disease conditions. These hub genes are important drivers of molecular networks in highly connected hubs within densely interacting genes, regulating critical pathways linked to CVD pathogeneses. ML classification models supported their potential as biomarkers for diagnostic applications with high accuracy (Soleymani et al., 2022). As pathway analyses are crucial for better comprehension of the complicated molecular landscape of CVDs (Patel et al., 2023), we performed functional analyses of the identified hub DEGs. Hub gene enrichment in KEGG maps and PPI networks revealed disruptions in the metabolic and signaling networks underlying the pathophysiology of CVD. Targeting these genes may have synergistic downstream effects due to their ability to integrate signals from multiple pathways simultaneously, including lipid metabolism (APOE, LDLR, PCSK9), renin-angiotensin system (ACE), TGF-β signaling (ACTA2), AMPK signaling (PRKAG2), cardiac muscle contraction (TNNT2) cascades.
As a key aim of this research, we also identified high-affinity binding ligands for the hub genes associated with CVD, as these can be utilized as biotargets for possible ameliorative strategies. In this regard, docking analysis identified the specific ligands among approved clinically relevant drugs as high-affinity binding ligands for the top DEGs. For this purpose, we only screened clinically approved drugs with known therapeutic actions for repurposition. Our results indicated that IL6 had a high binding affinity for evofolisat, in consistency with the anti- inflammatory actions of the latter. The interaction of TNF with rosuvastatin demonstrates its possible function in combating inflammation and lipid imbalance, consistent with evidence that statins decrease cardiovascular events. The binding of MYH6 with atenolol, and TNNT2 with omecamtiv mecarbil is consistent with the functions of beta-blockers and myosin activators in governing the contractility of the cardiac muscles and for heart failure treatment purposes. High-affinity interaction of LDLR and PCSK9 with ezetimibe and alirocumab supports cholesterol pathway drugs as potential therapeutics against CVDs (Soleymani et al., 2022). ACE’s interaction with lisinopril justifies the role of ACE inhibitors in the treatment of hypertension. In conclusion, hub genes identified in our study are not only crucial to the molecular foundations of CVDs but also provide opportunities for specialized treatment approaches. By enabling more accurate diagnoses, customized treatment regimens, and ultimately better patient outcomes, our data has the potential to completely transform the management of CVD.
5. Conclusions
Analyses of DEGs using public repositories of CVD subjects identified IL6, TNF, MYH6, APOE, LDLR, PCSK9, ACE, ACTA2, PRKAG2, and TNNT2 as the top ten features predicted among DEG biomarkers. Further, in silico analyses were performed to identify high-affinity binding drugs from amongst already clinically approved drugs against these biotargets. Our results revealed therapeutics that hold promise for safe, effective, and targeted treatment of CVDs. However, preclinical and clinical assessment of the identified chemicals is warrantied in future assessments.
Acknowledgments
This Project was funded by the Deanship of Scientific Research (DSR) at King Abdulaziz University, Jeddah, under grant no. (G:284- 828-1442). The authors, therefore, acknowledge with thanks DSR for technical and financial support.
CRediT authorship contribution statement
Hala Abubaker Bagabira: Conceptualization, data curation, writing, methodology, visualization, resources, software work, formal analysis, validation; Shimaa Mohammad Yousof: Conceptualization, data curation, writing, methodology, visualization, resources, software work, formal analysis, validation; Lamis Kaddam: Conceptualization, data curation, writing, methodology, visualization, resources, software work, formal analysis, validation; Mohamed A. Zayed: conceptualization, data curation, writing, methodology, visualization, resources, software work, formal analysis, validation; Sali Abubaker Bagabira: Conceptualization, data curation, methodology, visualization, resources, software work, formal analysis, validation, review and editing; Shafiul Haque: Conceptualization, data curation, writing, methodology, funding acquisition, visualization, supervision, resources, software work, formal analysis, validation, review and editing, Faraz Ahmad: Conceptualization, data curation, methodology, visualization, resources, software work, formal analysis, validation, review and editing, Sabiha Khatoone: Conceptualization, data curation, writing, methodology, visualization, supervision, resources, software work, formal analysis, validation, review and editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Declaration of Generative AI and AI-assisted technologies in the writing process
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Funding
Deanship of Scientific Research (DSR) at King Abdulaziz University, Jeddah, Saudi Arabia (Grant No. G:284-828-1442).
Supplementary data
Supplementary material to this article can be found online at https://dx.doi.org/10.25259/JKSUS_358_2024.
References
- Molecular docking as a tool for the discovery of molecular targets of nutraceuticals in diseases management. Sci. Rep.. 2023;13:13398. https://doi.org/10.1038/s41598-023-40160-2
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Artificial intelligence with multi-functional machine learning platform development for better healthcare and precision medicine. Database : The Journal of Biological Databases and Curation. 2020;2020:baaa010. https://doi.org/10.1093/database/baaa010
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- RNA-seq driven expression and enrichment analysis to investigate CVD genes with associated phenotypes among high-risk heart failure patients. Hum. Genomics. 2021;15:67. https://doi.org/10.1186/s40246-021-00367-8
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Application of support vector machine algorithm for early differential diagnosis of prostate cancer. Data Science and Management. 2023;6:1-12. https://doi.org/10.1016/j.dsm.2022.10.001
- [CrossRef] [Google Scholar]
- 10 Cardiovascular disease and risk management: Standards of medical care in diabetes-2022. Diabetes Care. 2022;45:S144-S174. https://doi.org/10.2337/dc22-S010
- [CrossRef] [PubMed] [Google Scholar]
- Machine learning analysis of RNA-seq data for diagnostic and prognostic prediction of colon cancer. Sensors (Basel, Switzerland). 2023;23:3080. https://doi.org/10.3390/s23063080
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Translating RNA sequencing into clinical diagnostics: Opportunities and challenges. Nat. Rev. Genet.. 2016;17:257-271. https://doi.org/10.1038/nrg.2016.10
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Transcriptome profiling in human diseases: New advances and perspectives. Int. J. Mol. Sci.. 2017;18:1652. https://doi.org/10.3390/ijms18081652
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The gene expression omnibus database. Methods in Molecular Biology (Clifton, N.J.). 2016;1418:93-110. https://doi.org/10.1007/978-1-4939-3578-9_5
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Machine learning in drug discovery: A review. Artificial Intelligence Review. 2022;55:1947-1999. https://doi.org/10.1007/s10462-021-10058-4
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Computational identification of differentially-expressed genes as suggested novel COVID-19 biomarkers: A bioinformatics analysis of expression profiles. Computational and Structural Biotechnology Journal. 2023;21:3339-3354. https://doi.org/10.1016/j.csbj.2023.06.007
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Multi-omics approaches for revealing the complexity of cardiovascular disease. Brief. Bioinform.. 2021;22:bbab061. https://doi.org/10.1093/bib/bbab061
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The application of big data to cardiovascular disease: Paths to precision medicine. J. Clin. Invest.. 2020;130:29-38. https://doi.org/10.1172/JCI129203
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Feature selection with the fisher score followed by the maximal clique centrality algorithm can accurately identify the hub genes of hepatocellular carcinoma. Sci. Rep.. 2019;9:17283. https://doi.org/10.1038/s41598-019-53471-0
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Protein–protein interaction network exploration using cytoscape. Protein-Protein Interactions 2023:419-427. https://doi.org/10.1007/978-1-0716-3327-4_32
- [Google Scholar]
- Cardiovascular Disease. StatPearls; 2023.
- Genomic approaches to identify and investigate genes associated with atrial fibrillation and heart failure susceptibility. Hum. Genomics. 2023;17:47. https://doi.org/10.1186/s40246-023-00498-0
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res.. 2015;43:e47. https://doi.org/10.1093/nar/gkv007
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Globalization, work, and cardiovascular disease. International Journal of Health Services : Planning, Administration, Evaluation. 2016;46:656-692. https://doi.org/10.1177/0020731416664687
- [CrossRef] [PubMed] [Google Scholar]
- Gene expression analysis of cardiovascular diseases. J. Am. Coll. Cardiol.. 2006;48:227-235. https://doi.org/10.1016/j.jacc.2006.02.070
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Protein–protein interaction prediction with deep learning: A comprehensive review. Computational and Structural Biotechnology Journal. 2022;20:5316-5341. https://doi.org/10.1016/j.csbj.2022.08.070
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Key topics in molecular docking for drug design. Int. J. Mol. Sci.. 2019;20:4574. https://doi.org/10.3390/ijms(2018)4574
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Emerging risk biomarkers in cardiovascular diseases and disorders. Journal of Lipids. 2015;2015:971453. https://doi.org/10.1155/(2015)/971453
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Artificial intelligence and machine learning approaches using gene expression and variant data for personalized medicine. Brief. Bioinform.. 2022;23:bbac191. https://doi.org/10.1093/bib/bbac191
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Novel biomarkers for cardiovascular risk prediction. Journal of Geriatric Cardiology: JGC. 2017;14:135-150. https://doi.org/10.11909/j.issn.1671-5411.2017.02.008
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Cardiovascular diseases (CVDs).
- ClusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Cambridge (Mass.). 2021;2:100141. https://doi.org/10.1016/j.xinn.2021.100141
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]




